

Lactate dehydrogenase B noncanonically promotes ferroptosis defense in KRAS-driven lung cancer
- PMID: 39643712
- PMCID: PMC11982314
- DOI: 10.1038/s41418-024-01427-x
Lactate dehydrogenase B noncanonically promotes ferroptosis defense in KRAS-driven lung cancer
Abstract
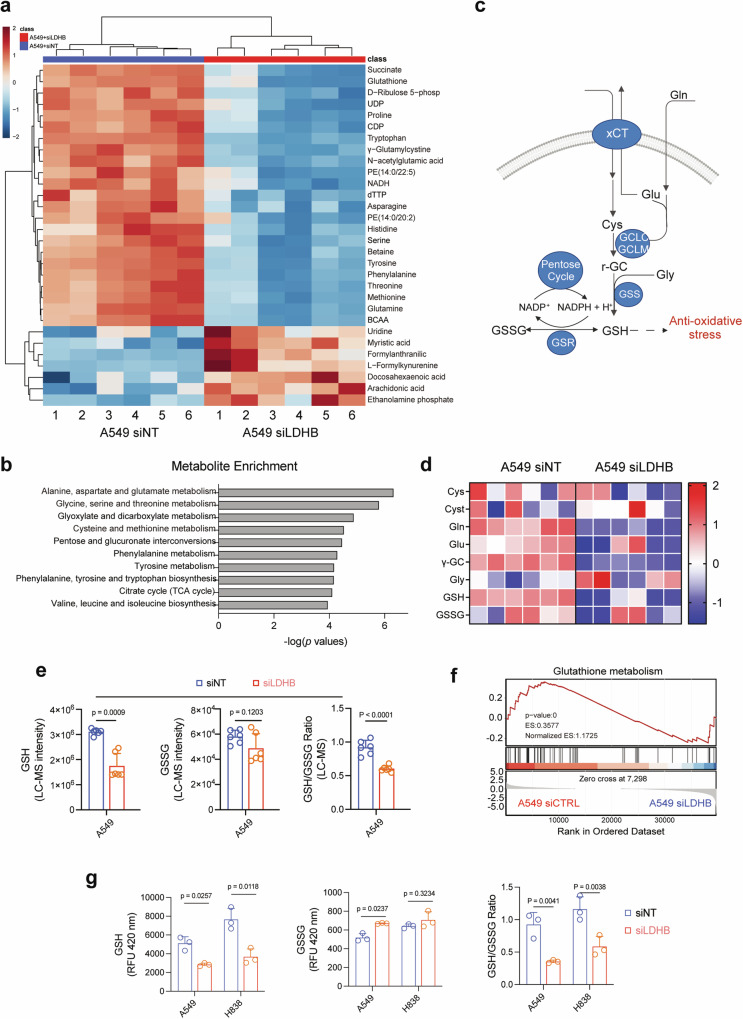
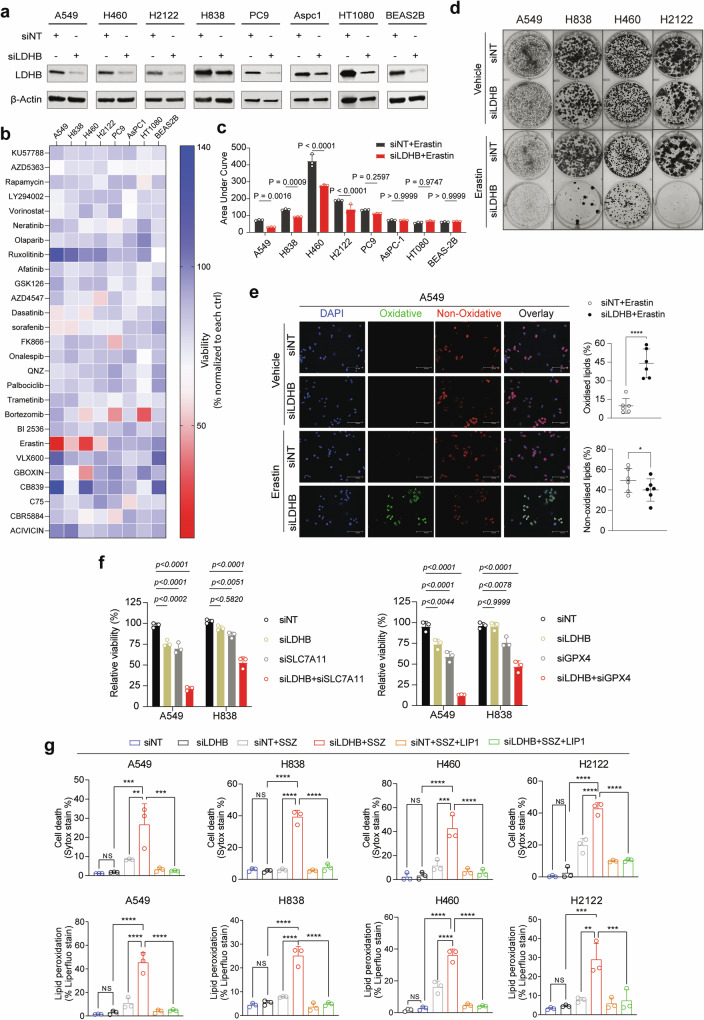
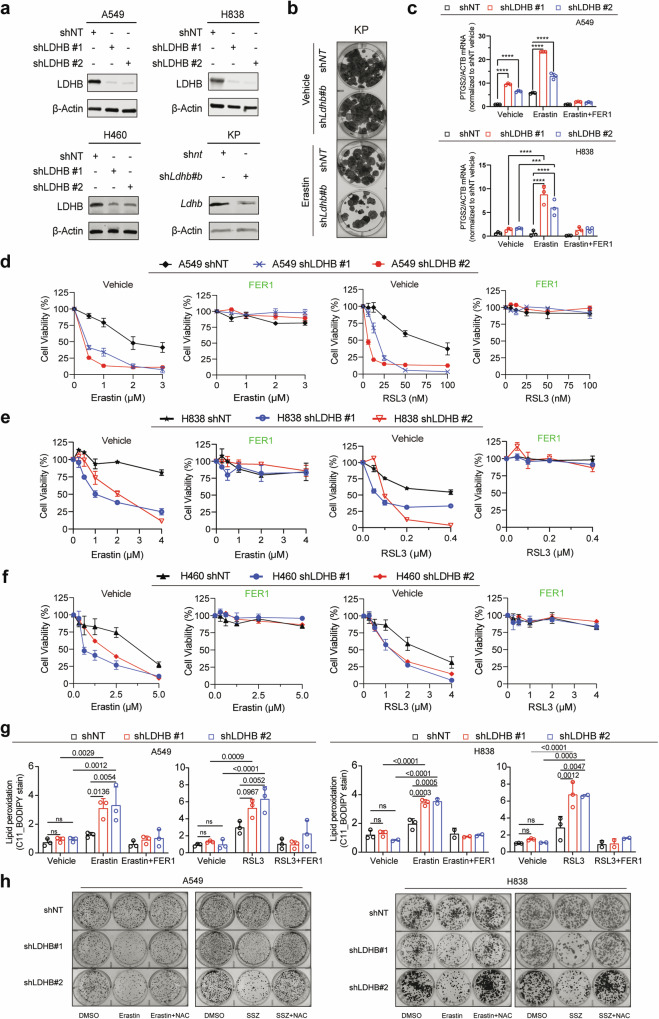
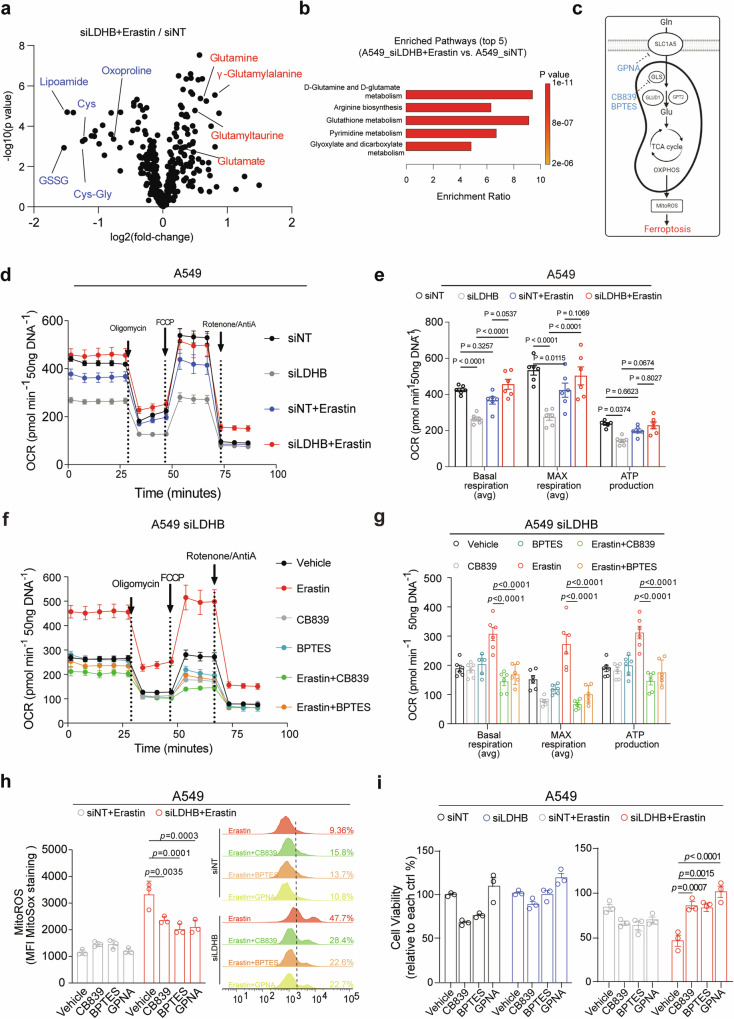
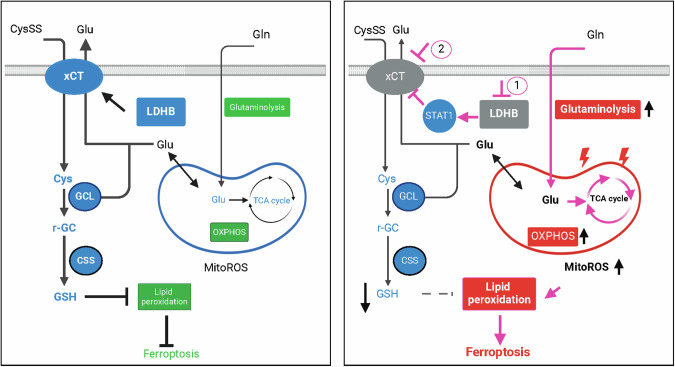
Ferroptosis is an oxidative, non-apoptotic cell death frequently inactivated in cancer, but the underlying mechanisms in oncogene-specific tumors remain poorly understood. Here, we discover that lactate dehydrogenase (LDH) B, but not the closely related LDHA, subunits of active LDH with a known function in glycolysis, noncanonically promotes ferroptosis defense in KRAS-driven lung cancer. Using murine models and human-derived tumor cell lines, we show that LDHB silencing impairs glutathione (GSH) levels and sensitizes cancer cells to blockade of either GSH biosynthesis or utilization by unleashing KRAS-specific, ferroptosis-catalyzed metabolic synthetic lethality, culminating in increased glutamine metabolism, oxidative phosphorylation (OXPHOS) and mitochondrial reactive oxygen species (mitoROS). We further show that LDHB suppression upregulates STAT1, a negative regulator of SLC7A11, thereby reducing SLC7A11-dependent GSH metabolism. Our study uncovers a previously undefined mechanism of ferroptosis resistance involving LDH isoenzymes and provides a novel rationale for exploiting oncogene-specific ferroptosis susceptibility to treat KRAS-driven lung cancer.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: Mouse studies were approved by the Veterinary Office of the Canton Bern, Switzerland, and conducted in accordance with Institutional Animal Care.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
