

This is a preprint.
Engineered CRISPR-Base Editors as a Permanent Treatment for Familial Dysautonomia
- PMID: 39651221
- PMCID: PMC11623606
- DOI: 10.1101/2024.11.27.625322
Engineered CRISPR-Base Editors as a Permanent Treatment for Familial Dysautonomia
Abstract
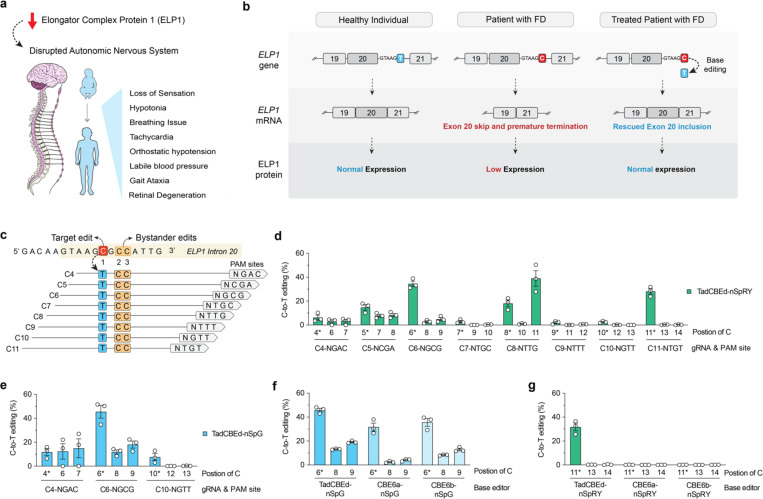
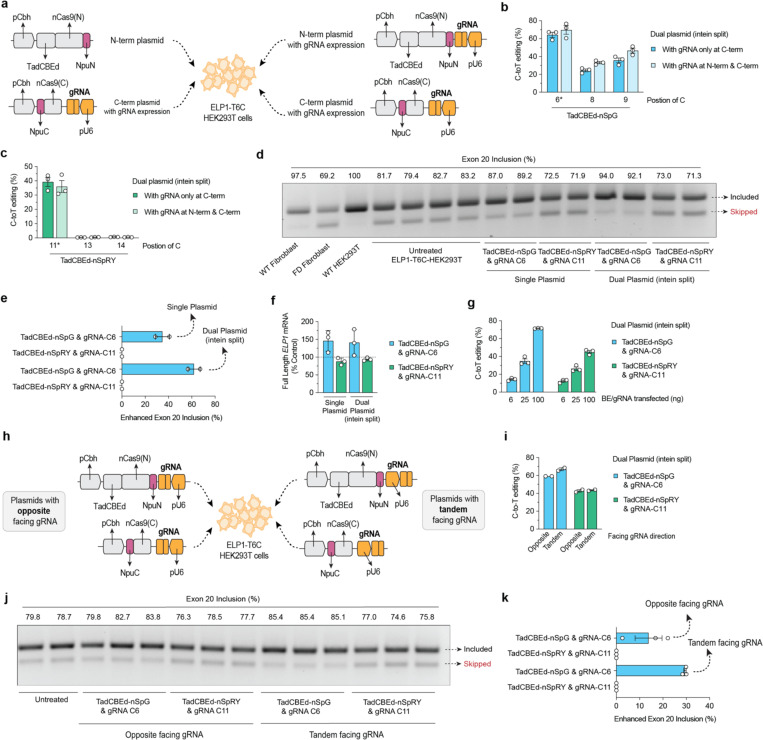
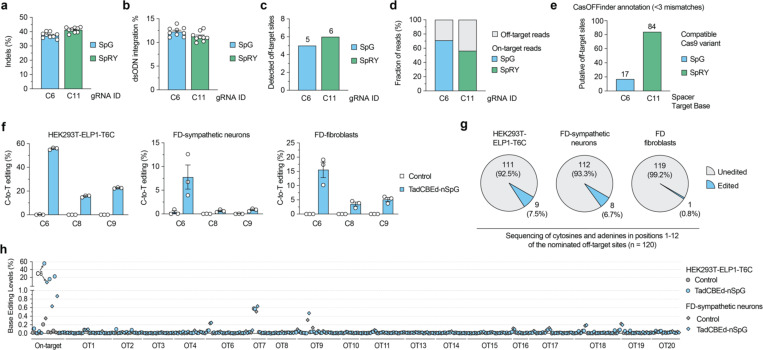
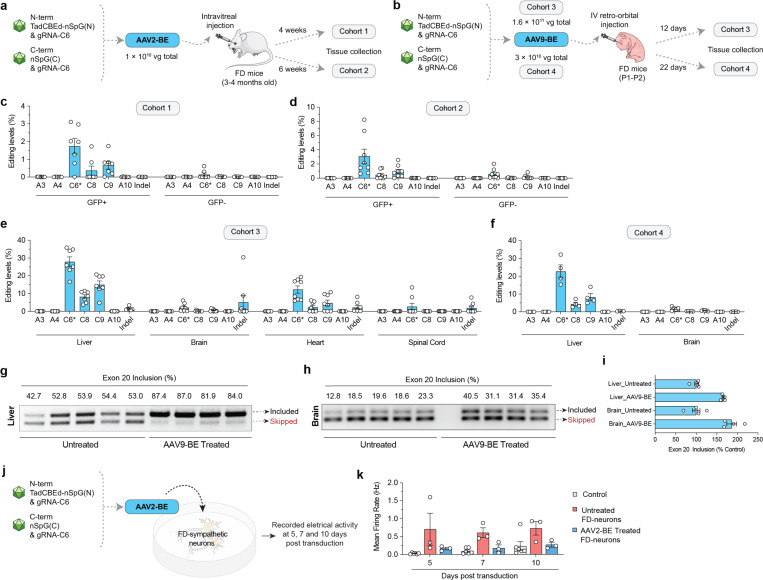
Familial dysautonomia (FD) is a fatal autosomal recessive congenital neuropathy caused by a T-to-C mutation in intron 20 of the Elongator acetyltransferase complex subunit 1 (ELP1) gene, which causes tissue-specific skipping of exon 20 and reduction of ELP1 protein. Here, we developed a base editor (BE) approach to precisely correct this mutation. By optimizing Cas9 variants and screening multiple gRNAs, we identified a combination that was able to promote up to 70% on-target editing in HEK293T cells harboring the ELP1 T-to-C mutation. These editing levels were sufficient to restore exon 20 inclusion in the ELP1 transcript. Moreover, we optimized an engineered dual intein-split system to deliver these constructs in vivo. Mediated by adeno-associated virus (AAV) delivery, this BE strategy effectively corrected the liver and brain ELP1 splicing defects in a humanized FD mouse model carrying the ELP1 T-to-C mutation and rescued the FD phenotype in iPSC-derived sympathetic neurons. Importantly, we observed minimal off-target editing demonstrating high levels of specificity with these optimized base editors. These findings establish a novel and highly precise BE-based therapeutic approach to correct the FD mutation and associated splicing defects and provide the foundation for the development of a transformative, permanent treatment for this devastating disease.
Keywords: CRISPR; Cas9; ELP1; bystander editing; genome editing; neurodegenerative disease; splicing.
Conflict of interest statement
Competing interests B.P.K., E.M. and C.R.R.A are inventors on a patent application filed by Mass General Brigham (MGB) that describes genome editing technologies to treat FD. B.P.K. and C.R.R.A. are inventors on additional patents or patent applications filed by MGB that describe genome engineering technologies, including to treat SMA. S.A.S. is an inventor on several U.S. and foreign patents and patent applications assigned to the Massachusetts General Hospital, including U.S Patents 8,729,025 and 9,265,766, both entitled “Methods for altering mRNA splicing and treating familial dysautonomia by administering benzyladenine,” filed on August 31, 2012 and May 19, 2014 and related to use of kinetin; and U.S. Patent 10,675,475 entitled, “Compounds for improving mRNA splicing” filed on July 14, 2017 and related to use of BPN-15477. E.M. and S.A.S. are inventors on an International Patent Application Number PCT/US2021/012103, assigned to Massachusetts General Hospital and entitled “ RNA Splicing Modulation” related to use of BPN-15477 in modulating splicing.B.P.K. is a consultant for EcoR1 capital, Novartis Venture Fund, and Jumble Therapeutics, and is on the scientific advisory boards of Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. E.M. is a consultant for ReviR Therapeutics. C.R.R.A is a consultant for Ilios Therapeutics and Biogen and holds stocks in publicly traded companies developing gene therapies. B.P.K. and C.R.R.A interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. The other authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources