

Multiplexing cortical brain organoids for the longitudinal dissection of developmental traits at single-cell resolution
- PMID: 39653820
- PMCID: PMC11810796
- DOI: 10.1038/s41592-024-02555-5
Multiplexing cortical brain organoids for the longitudinal dissection of developmental traits at single-cell resolution
Abstract
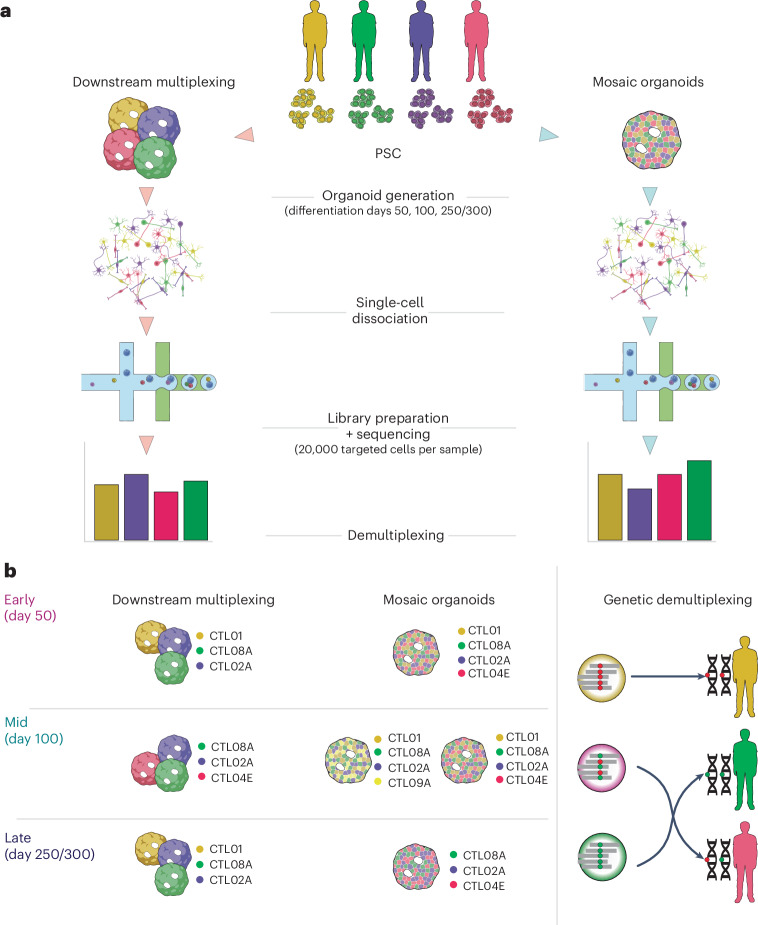
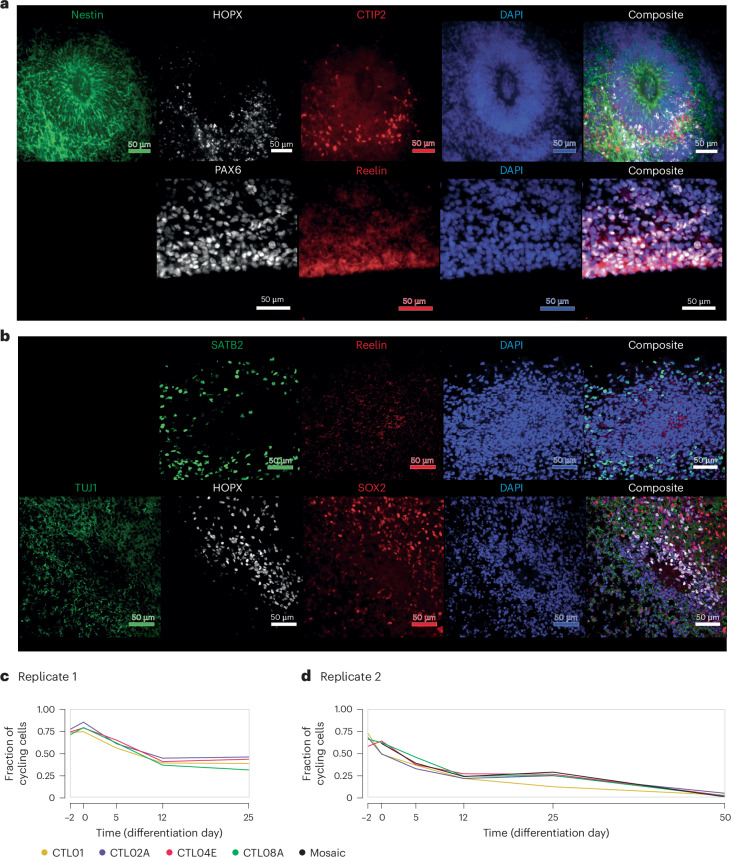
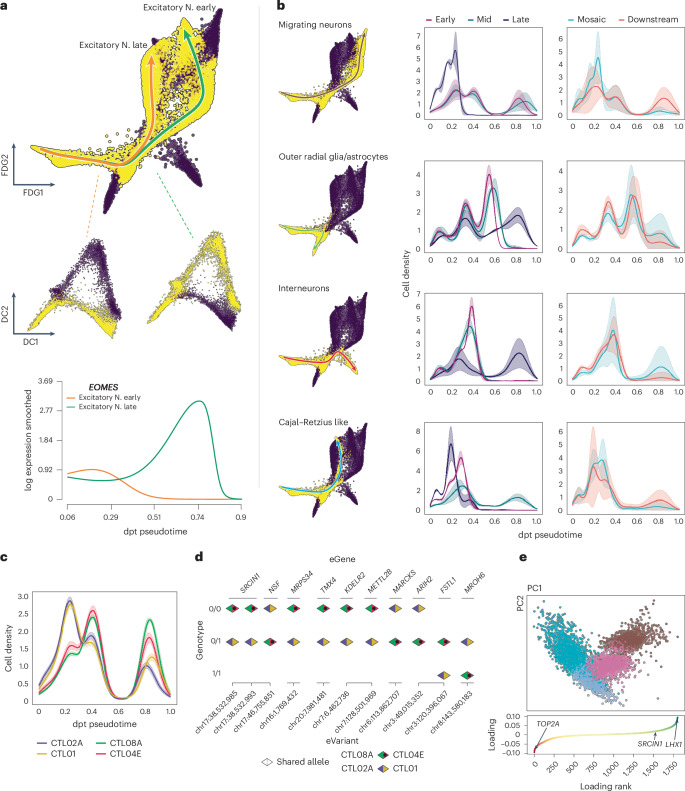
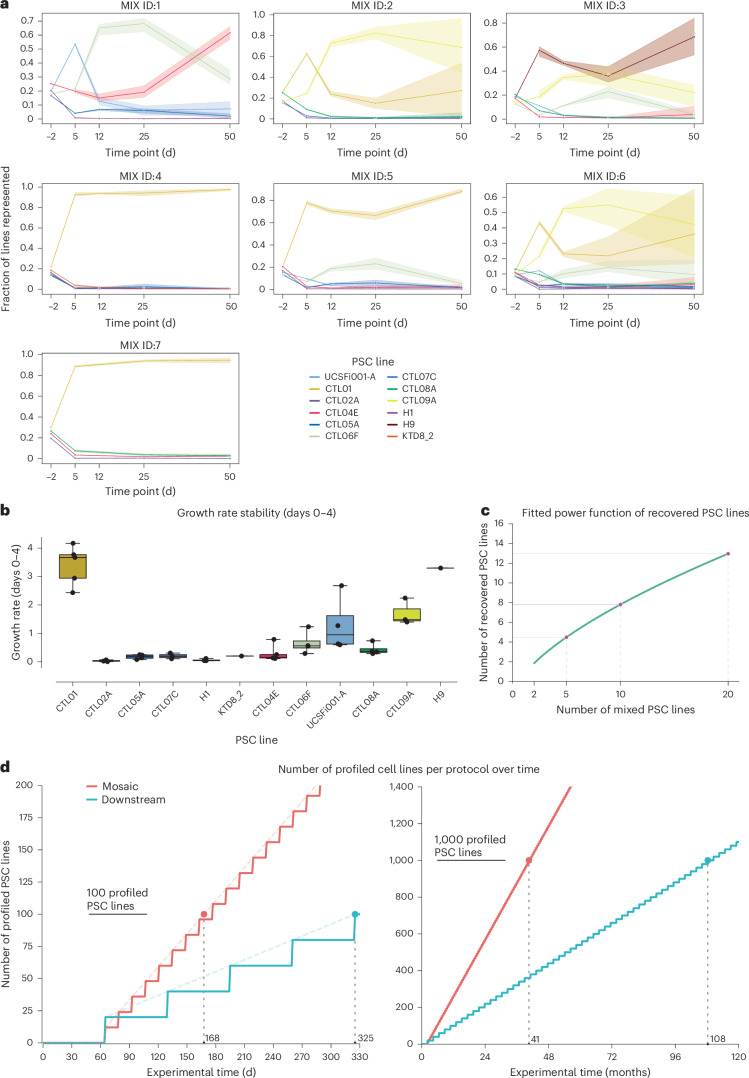
Dissecting human neurobiology at high resolution and with mechanistic precision requires a major leap in scalability, given the need for experimental designs that include multiple individuals and, prospectively, population cohorts. To lay the foundation for this, we have developed and benchmarked complementary strategies to multiplex brain organoids by pooling cells from different pluripotent stem cell (PSC) lines either during organoid generation (mosaic models) or before single-cell RNA sequencing (scRNA-seq) library preparation (downstream multiplexing). We have also developed a new computational method, SCanSNP, and a consensus call to deconvolve cell identities, overcoming current criticalities in doublets and low-quality cell identification. We validated both multiplexing methods for charting neurodevelopmental trajectories at high resolution, thus linking specific individuals' trajectories to genetic variation. Finally, we modeled their scalability across different multiplexing combinations and showed that mosaic organoids represent an enabling method for high-throughput settings. Together, this multiplexing suite of experimental and computational methods provides a highly scalable resource for brain disease and neurodiversity modeling.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: F.J.T. consults for Immunai, Singularity Bio, CytoReason and Omniscope and has ownership interest in Dermagnostix and Cellarity. The other authors declare no competing interests.
Figures












References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
