

Structure-based drug design with equivariant diffusion models
- PMID: 39653846
- PMCID: PMC11659159
- DOI: 10.1038/s43588-024-00737-x
Structure-based drug design with equivariant diffusion models
Abstract
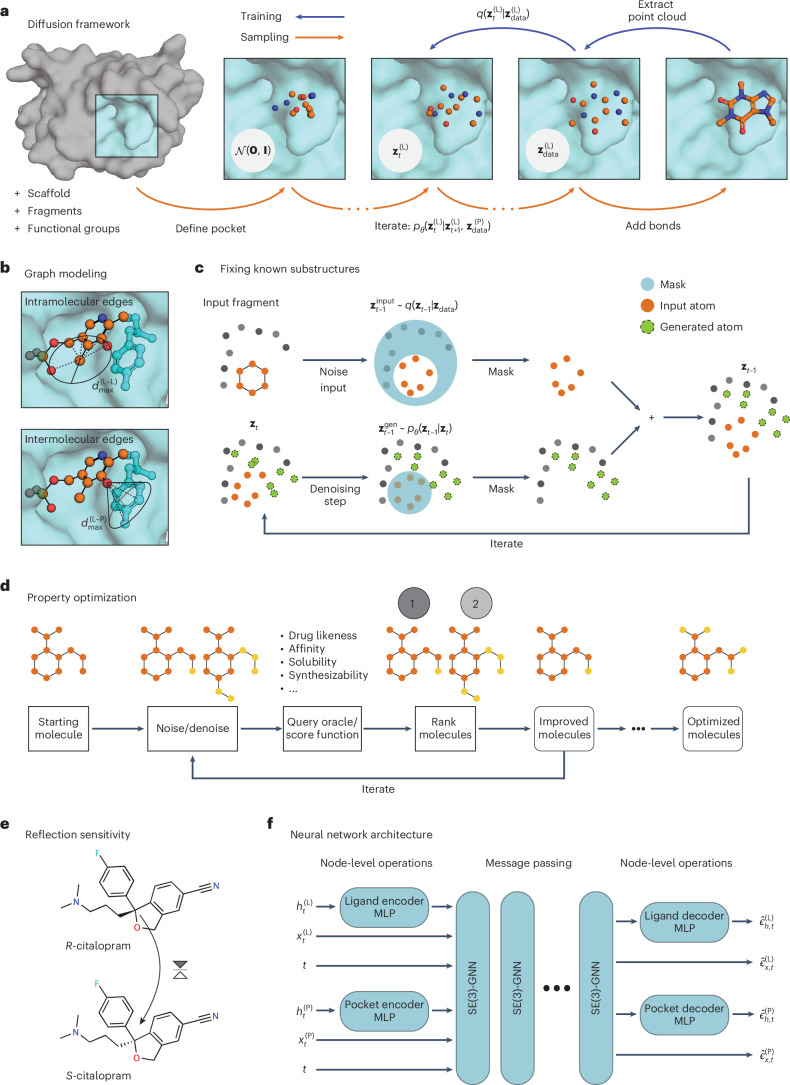
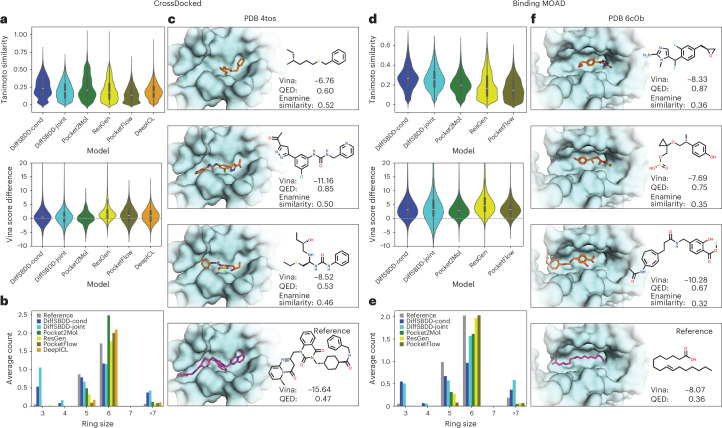
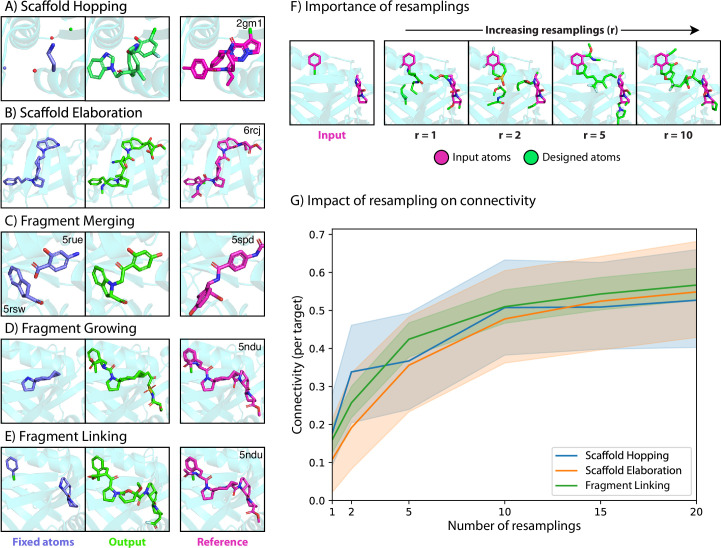
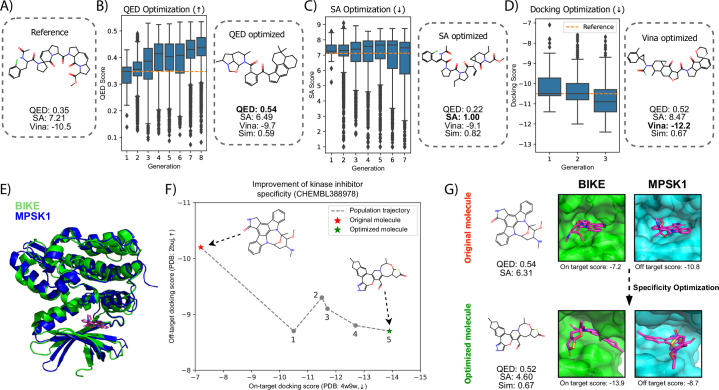
Structure-based drug design (SBDD) aims to design small-molecule ligands that bind with high affinity and specificity to pre-determined protein targets. Generative SBDD methods leverage structural data of drugs with their protein targets to propose new drug candidates. However, most existing methods focus exclusively on bottom-up de novo design of compounds or tackle other drug development challenges with task-specific models. The latter requires curation of suitable datasets, careful engineering of the models and retraining from scratch for each task. Here we show how a single pretrained diffusion model can be applied to a broader range of problems, such as off-the-shelf property optimization, explicit negative design and partial molecular design with inpainting. We formulate SBDD as a three-dimensional conditional generation problem and present DiffSBDD, an SE(3)-equivariant diffusion model that generates novel ligands conditioned on protein pockets. Furthermore, we show how additional constraints can be used to improve the generated drug candidates according to a variety of computational metrics.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- 716058/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 10030 188744/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
LinkOut - more resources
Full Text Sources
Other Literature Sources
