

BICEP: Bayesian inference for rare genomic variant causality evaluation in pedigrees
- PMID: 39656772
- PMCID: PMC11645550
- DOI: 10.1093/bib/bbae624
BICEP: Bayesian inference for rare genomic variant causality evaluation in pedigrees
Abstract
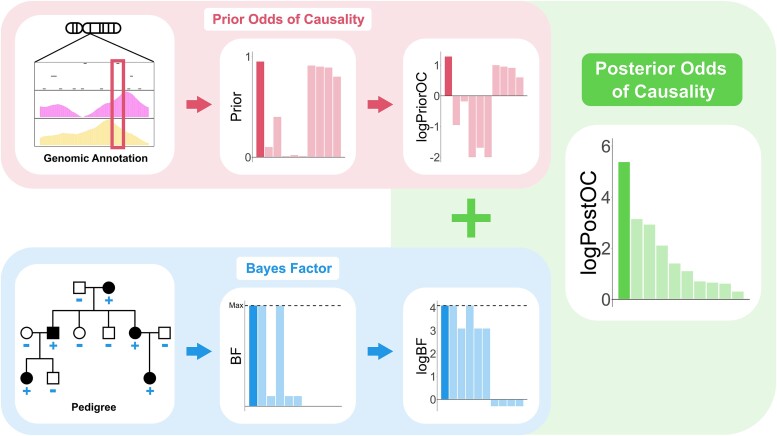
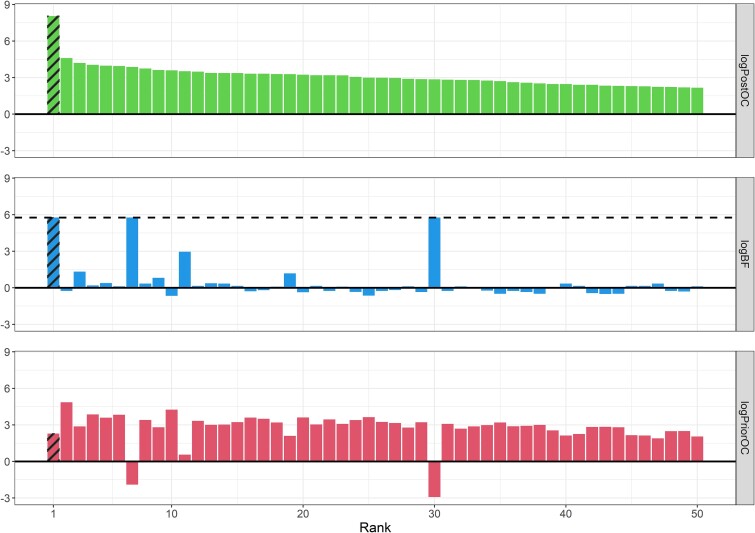
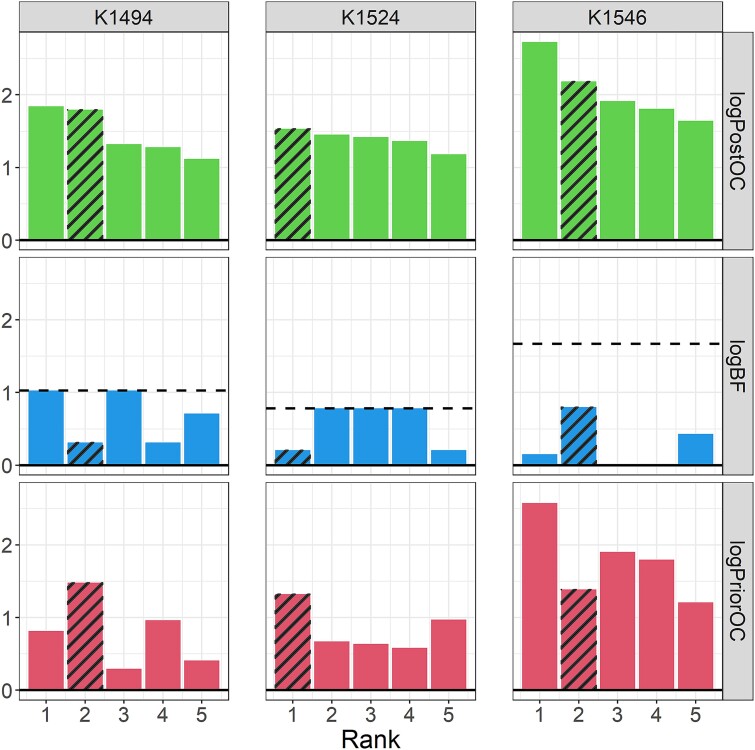
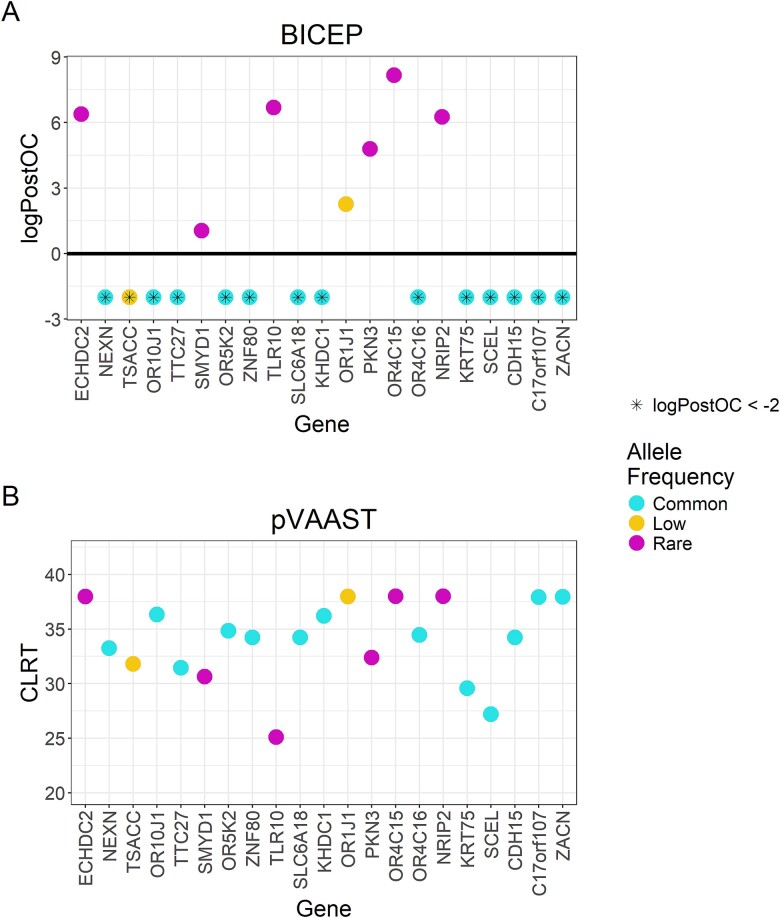
Next-generation sequencing is widely applied to the investigation of pedigree data for gene discovery. However, identifying plausible disease-causing variants within a robust statistical framework is challenging. Here, we introduce BICEP: a Bayesian inference tool for rare variant causality evaluation in pedigree-based cohorts. BICEP calculates the posterior odds that a genomic variant is causal for a phenotype based on the variant cosegregation as well as a priori evidence such as deleteriousness and functional consequence. BICEP can correctly identify causal variants for phenotypes with both Mendelian and complex genetic architectures, outperforming existing methodologies. Additionally, BICEP can correctly down-weight common variants that are unlikely to be involved in phenotypic liability in the context of a pedigree, even if they have reasonable cosegregation patterns. The output metrics from BICEP allow for the quantitative comparison of variant causality within and across pedigrees, which is not possible with existing approaches.
Keywords: Bayes factor; Bayesian inference; next-generation sequencing; pedigree; posterior odds of causality; prior odds of causality.
© The Author(s) 2024. Published by Oxford University Press.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
