

Genomic virulence markers are associated with severe outcomes in patients with Pseudomonas aeruginosa bloodstream infection
- PMID: 39663376
- PMCID: PMC11634891
- DOI: 10.1038/s43856-024-00696-4
Genomic virulence markers are associated with severe outcomes in patients with Pseudomonas aeruginosa bloodstream infection
Abstract
Background: Pseudomonas aeruginosa (PA) bloodstream infection (BSI) is a common healthcare-associated complication linked to antimicrobial resistance and high mortality. Ongoing clinical trials are exploring novel anti-virulence agents, yet studies on how bacterial virulence affects PA infection outcomes is conflicting and data from real-world clinical populations is limited.
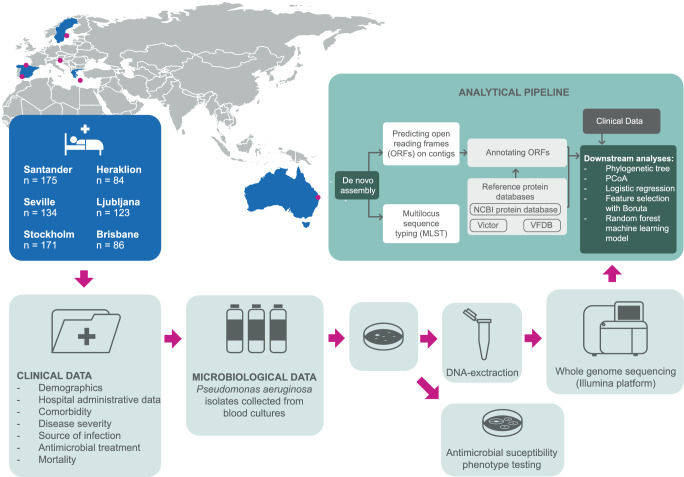
Methods: We studied a multicentre cohort of 773 adult patients with PA BSI consecutively collected during 7-years from sites in Europe and Australia. Comprehensive clinical data and whole-genome sequencing of all bacterial strains were obtained.
Results: Based on the virulence genotype, we identify several virulence clusters, each showing varying proportions of multidrug-resistant phenotypes. Genes tied to biofilm synthesis and epidemic clones ST175 and ST235 are associated with mortality, while the type III secretion system is associated with septic shock. Adding genomic biomarkers to machine learning models based on clinical data indicates improved prediction of severe outcomes in PA BSI patients.
Conclusions: These findings suggest that virulence markers provide prognostic information with potential applications in guiding adjuvant sepsis treatments.
Plain language summary
Pseudomonas aeruginosa bacteria are often found in the hospital environment, primarily infecting vulnerable patients with underlying health conditions. Due to antibiotic resistance, which occurs when bacteria are not killed by antibiotic treatment, these infections are often difficult to treat, and death rates are high. In this study, we analyzed data from patients in Europe and Australia with bloodstream infections to understand how bacterial traits affect patient outcomes. Using genetic information from the bacteria, we identified characteristics associated with antibiotic resistance. In addition, we found certain bacterial traits, such as the ability to synthesize toxins and biofilms, were linked to disease severity and mortality risk. These findings indicate that specific characteristics of P. aeruginosa may influence the severity of infection and could be targeted in newly developed treatments.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare the following competing interests: L.M.-M. reports receiving grants from Shionogi, Merck Sharp, and Dohme, and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Pfizer, hionogi, Merck Sharp, and Dohme. No other authors report any conflict of interest.
Figures





References
-
- Wenzel, R. P. et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 Cases from a Prospective Nationwide Surveillance Study. Clin. Infect. Dis.39, 309–317 (2004). - PubMed
-
- Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis.18, 318–327 (2018). - PubMed
-
- Parkins, M. D., Gregson, D. B., Pitout, J. D. D., Ross, T. & Laupland, K. B. Population-based study of the epidemiology and the risk factors for Pseudomonas aeruginosa bloodstream infection. Infection38, 25–32 (2010). - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
