

Central control of dynamic gene circuits governs T cell rest and activation
- PMID: 39663454
- PMCID: PMC11754113
- DOI: 10.1038/s41586-024-08314-y
Central control of dynamic gene circuits governs T cell rest and activation
Abstract
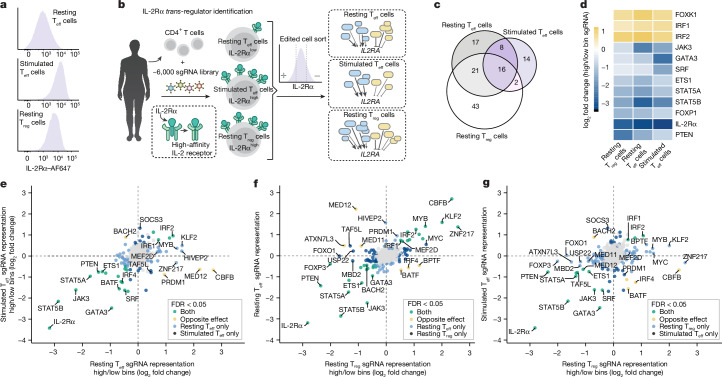
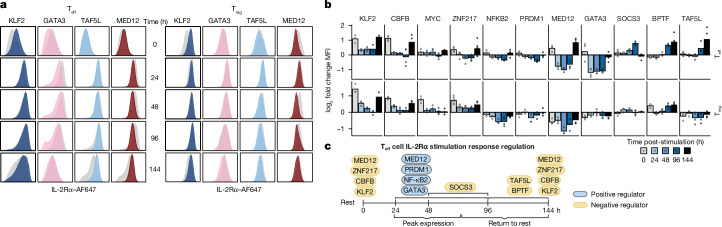
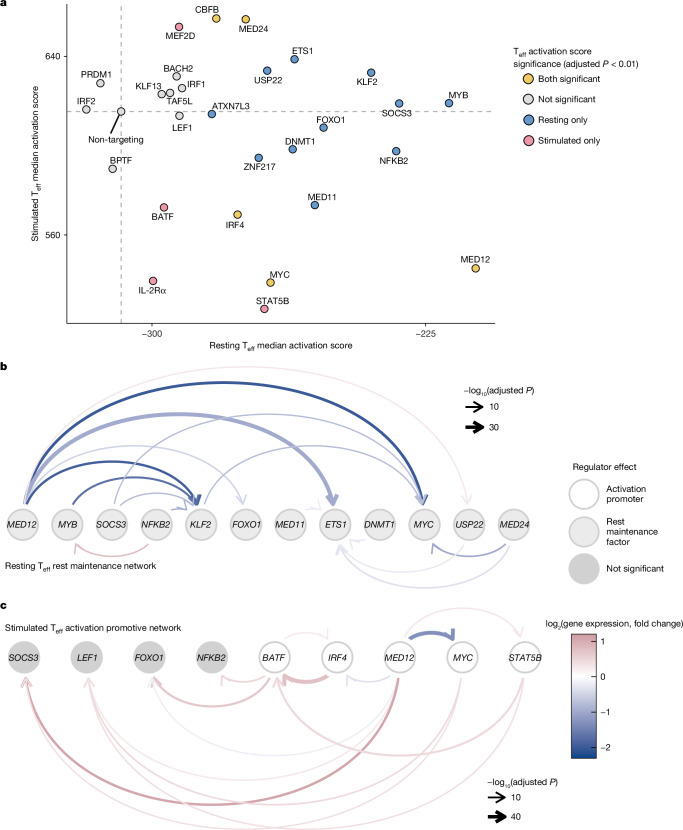
The ability of cells to maintain distinct identities and respond to transient environmental signals requires tightly controlled regulation of gene networks1-3. These dynamic regulatory circuits that respond to extracellular cues in primary human cells remain poorly defined. The need for context-dependent regulation is prominent in T cells, where distinct lineages must respond to diverse signals to mount effective immune responses and maintain homeostasis4-8. Here we performed CRISPR screens in multiple primary human CD4+ T cell contexts to identify regulators that control expression of IL-2Rα, a canonical marker of T cell activation transiently expressed by pro-inflammatory effector T cells and constitutively expressed by anti-inflammatory regulatory T cells where it is required for fitness9-11. Approximately 90% of identified regulators of IL-2Rα had effects that varied across cell types and/or stimulation states, including a subset that even had opposite effects across conditions. Using single-cell transcriptomics after pooled perturbation of context-specific screen hits, we characterized specific factors as regulators of overall rest or activation and constructed state-specific regulatory networks. MED12 - a component of the Mediator complex - serves as a dynamic orchestrator of key regulators, controlling expression of distinct sets of regulators in different T cell contexts. Immunoprecipitation-mass spectrometry revealed that MED12 interacts with the histone methylating COMPASS complex. MED12 was required for histone methylation and expression of genes encoding key context-specific regulators, including the rest maintenance factor KLF2 and the versatile regulator MYC. CRISPR ablation of MED12 blunted the cell-state transitions between rest and activation and protected from activation-induced cell death. Overall, this work leverages CRISPR screens performed across conditions to define dynamic gene circuits required to establish resting and activated T cell states.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: A.M. is a cofounder of Site Tx, Arsenal Biosciences, Spotlight Therapeutics and Survey Genomics; serves on the boards of directors at Site Tx, Spotlight Therapeutics and Survey Genomics; is a member of the scientific advisory boards of Site Tx, Arsenal Biosciences, Cellanome, Spotlight Therapeutics, Survey Genomics, NewLimit, Amgen and Tenaya; owns stock in Arsenal Biosciences, Site Tx, Cellanome, Spotlight Therapeutics, NewLimit, Survey Genomics, Tenaya and Lightcast; has received fees from Site Tx, Arsenal Biosciences, Cellanome, Spotlight Therapeutics, NewLimit, Gilead, Pfizer, 23andMe, PACT Pharma, Juno Therapeutics, Tenaya, Lightcast, Trizell, Vertex, Merck, Amgen, Genentech, GLG, ClearView Healthcare, AlphaSights, Rupert Case Management, Bernstein and ALDA; is an investor in and informal advisor to Offline Ventures; and a client of EPIQ. The Marson laboratory has received research support from the Parker Institute for Cancer Immunotherapy, the Emerson Collective, Arc Institute, Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead and Anthem and reagents from Genscript and Illumina. The Krogan Laboratory has received research support from Vir Biotechnology, F. Hoffmann-La Roche and Rezo Therapeutics. N.J.K. has a financially compensated consulting agreement with Maze Therapeutics. N.J.K. is the President and on the Board of Directors of Rezo Therapeutics; and is a shareholder in Tenaya Therapeutics, Maze Therapeutics, Rezo Therapeutics, GEn1E Lifesciences and Interline Therapeutics. J.W.F. was a consultant for NewLimit; is an employee of Genentech; and has equity in Roche. A.T.S. is a founder of Immunai, Cartography Biosciences, Santa Ana Bio and Prox Biosciences; is an advisor to Zafrens and Wing Venture Capital; and receives research funding from Astellas and Merck Research Laboratories. Patent applications have been filed based on the findings described here. The other authors declare no competing interests.
Figures













References
-
- Ota, M. et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell184, 3006–3021.e17 (2021). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
