

Predisposition Footprints in the Somatic Genome of Wilms Tumors
- PMID: 39665570
- PMCID: PMC7617291
- DOI: 10.1158/2159-8290.CD-24-0878
Predisposition Footprints in the Somatic Genome of Wilms Tumors
Abstract
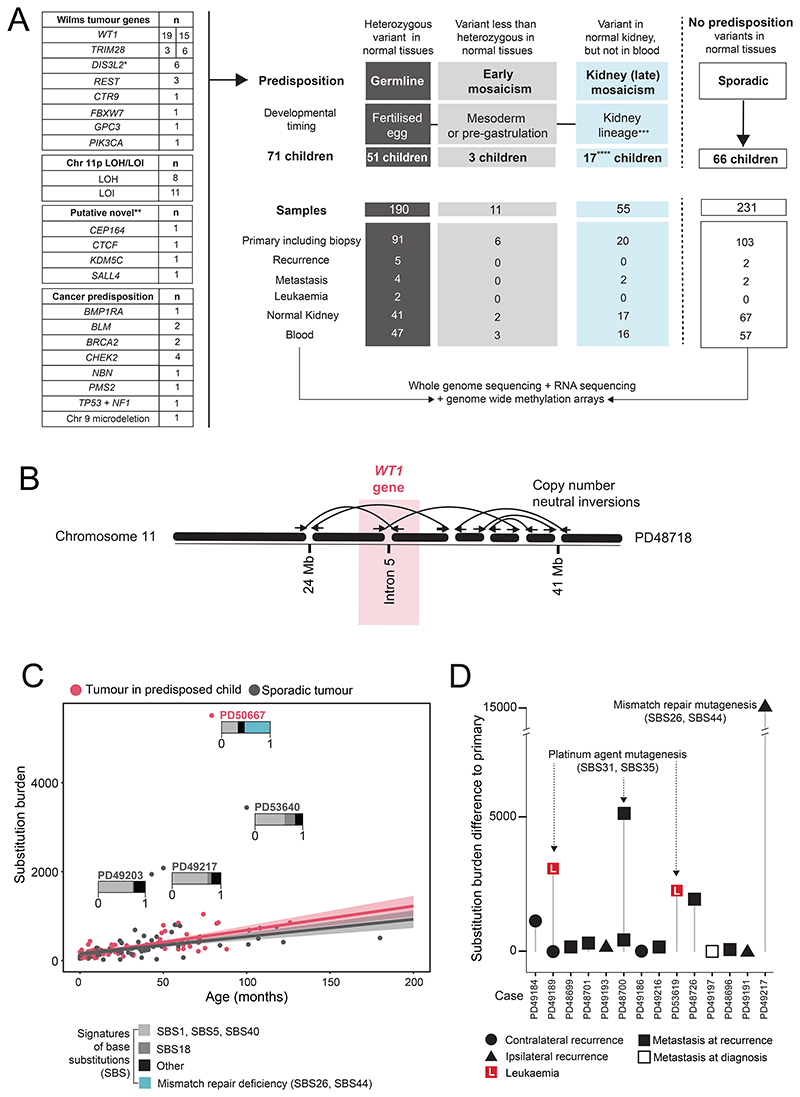
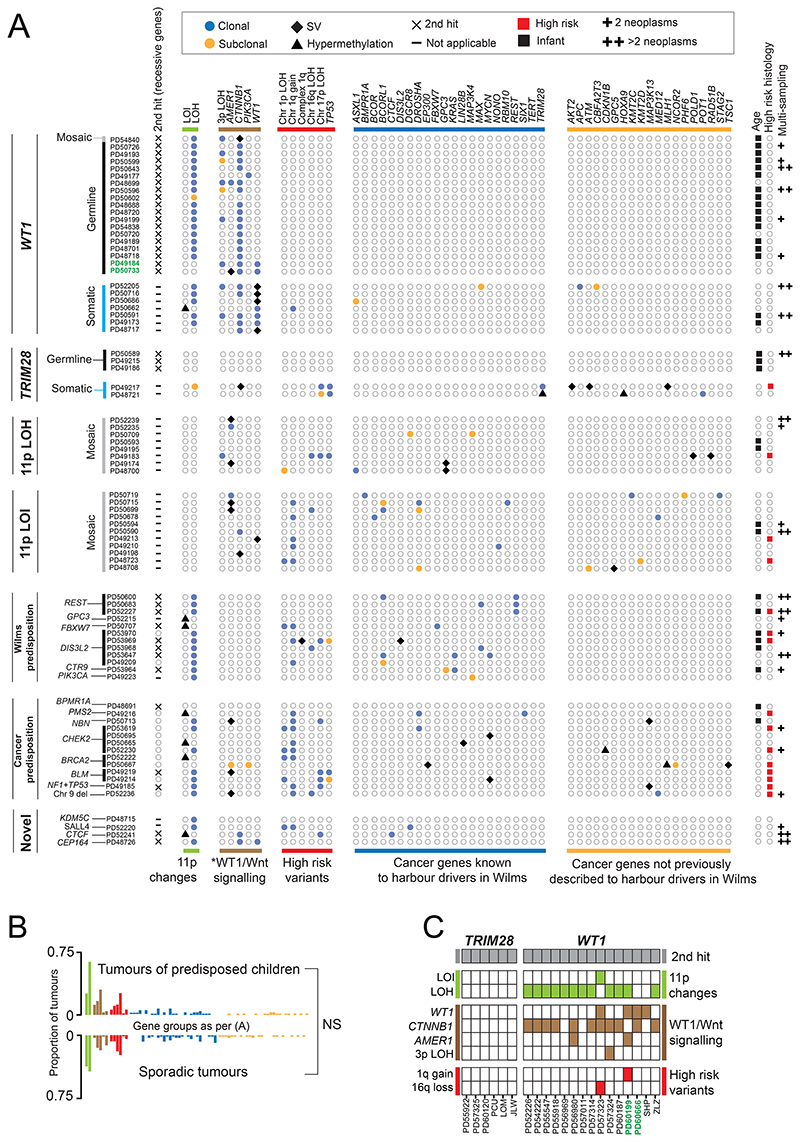
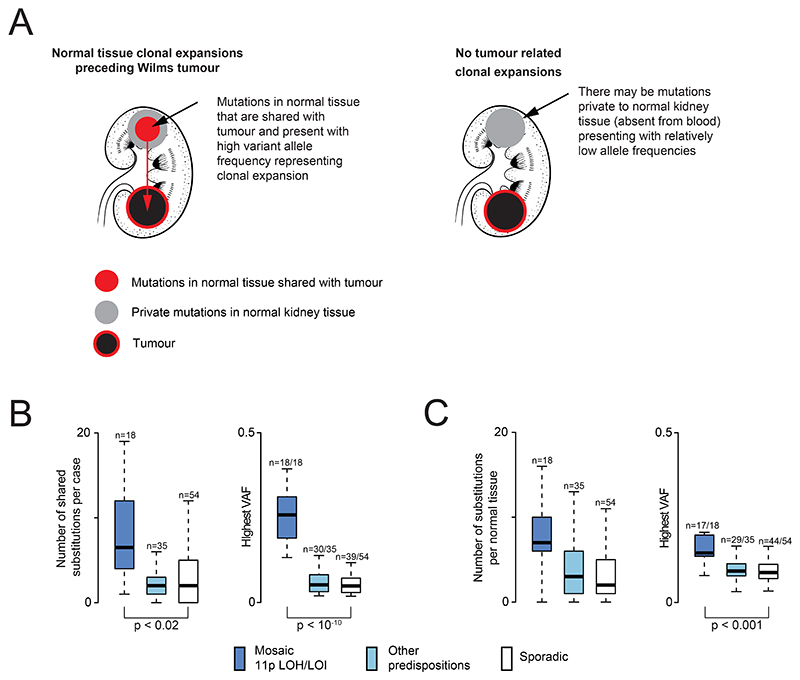
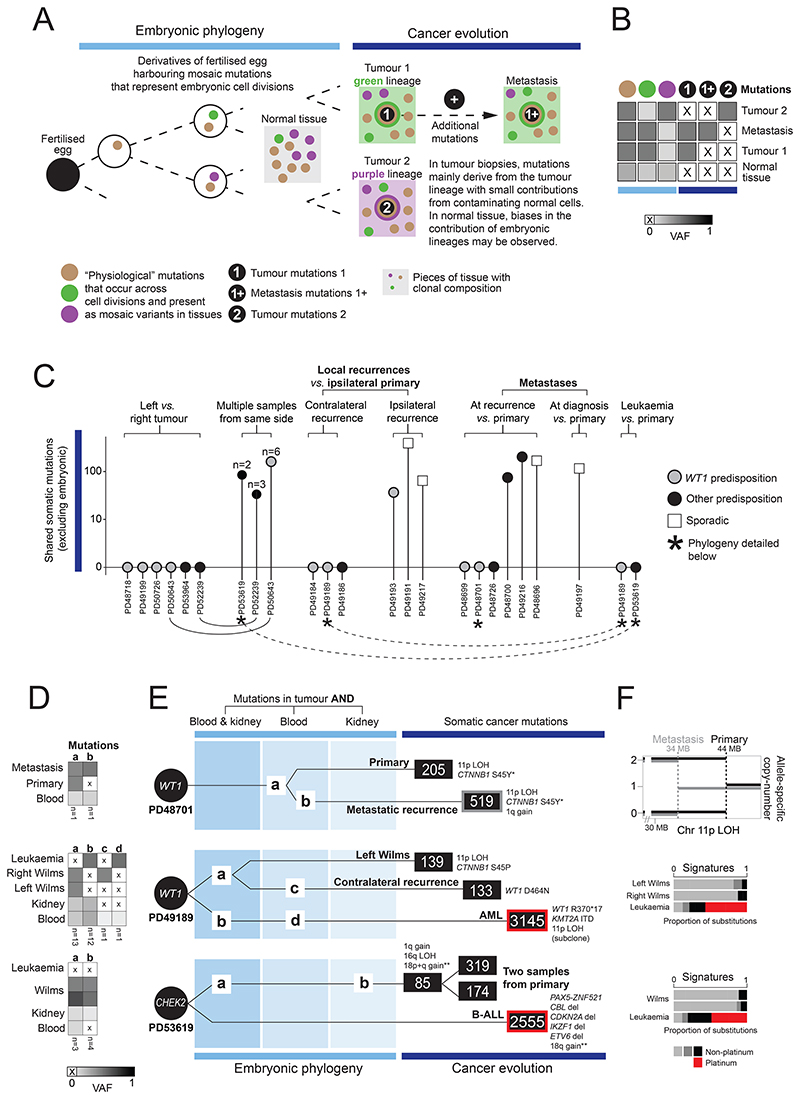
Approximately 10% of children with cancer harbor a mutation in a predisposition gene. In children with the kidney cancer Wilms tumor, the prevalence is as high as 30%. Certain predispositions are associated with defined histological and clinical features, suggesting differences in tumorigenesis. To investigate this, we assembled a cohort of 137 children with Wilms tumor, of whom 71 had a pathogenic germline or mosaic variant. We examined 237 neoplasms (including two secondary leukemias), utilizing whole-genome sequencing, RNA sequencing, and genome-wide methylation, validating our findings in an independent cohort. Tumor development differed in children harboring a predisposition, depending on the variant gene and its developmental timing. Differences pervaded the repertoire of driver events, including high-risk mutations, the clonal architecture of normal kidneys, and the relatedness of neoplasms from the same individual. Our findings indicate that predisposition may preordain Wilms tumorigenesis, suggesting a variant-specific approach to managing children merits consideration. Significance: Tumors that arise in children with a cancer predisposition may develop through the same mutational pathways as sporadic tumors. We examined this question in the childhood kidney cancer, Wilms tumor. We found that certain predispositions dictate the genetic development of tumors, with clinical implications for these children. See related commentary by Brzezinski and Malkin, p. 258.
©2025 American Association for Cancer Research.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
References
-
- Kratz CP, Jongmans MC, Cavé H, Wimmer K, Behjati S, Guerrini-Rousseau L, et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc Health. 2021;5:142–54. - PubMed
-
- Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Nat Med. Vol. 26. Springer Science and Business Media LLC; 2020. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer; pp. 1742–53. - PubMed
-
- Hol JA, Kuiper RP, van Dijk F, Waanders E, van Peer SE, Koudijs MJ, et al. Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J Clin Oncol. 2022;40:1892–902. doi: 10.1200/JCO.21.02510. - DOI - PMC - PubMed
MeSH terms
Grants and funding
- Wenner-Gren Stiftelserna (Wenner-Gren Foundations)
- 223135/WT_/Wellcome Trust/United Kingdom
- Children's Kidney Care Fund/University of Cambridge (cambridgeuniversity)
- Little Princess Trust (LPT)
- 50-2709-Gr2/Deutsche Krebshilfe (German Cancer Aid)
- Wilhelm Sander-Stiftung (Wilhelm Sander Foundation)
- NIHR203312/NIHR Cambridge Biomedical Research Centre (NIHR Cambridge BRC)
- 223135/Z/21/Z/Wellcome Trust (WT)
- 220540/WT_/Wellcome Trust/United Kingdom
- NIHR Great Ormond Street Hospital Biomedical Research Centre (BRC)
- 220540/Z/20/A/Wellcome Trust (WT)
- NIHR203312/DH_/Department of Health/United Kingdom
- Ge539/Deutsche Forschungsgemeinschaft (DFG)
LinkOut - more resources
Full Text Sources
Medical
