

Baseline colitogenicity and acute perturbations of gut microbiota in immunotherapy-related colitis
- PMID: 39666007
- PMCID: PMC11636624
- DOI: 10.1084/jem.20232079
Baseline colitogenicity and acute perturbations of gut microbiota in immunotherapy-related colitis
Abstract
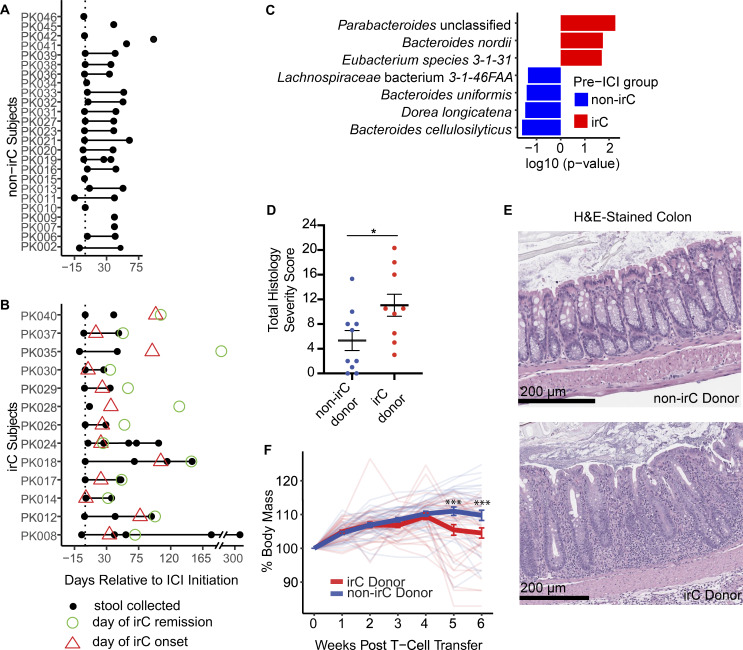
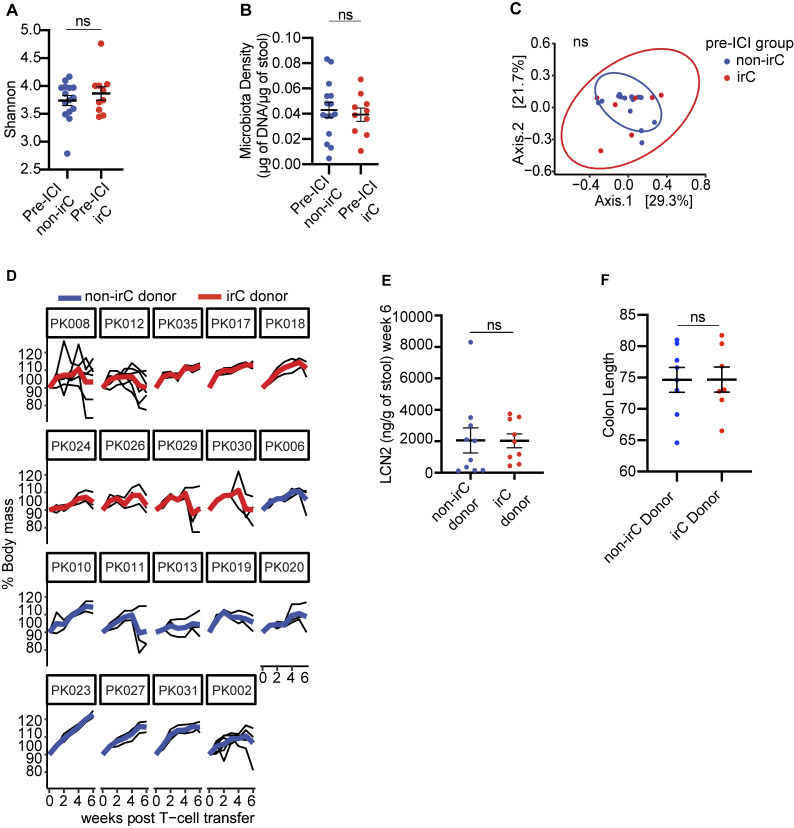
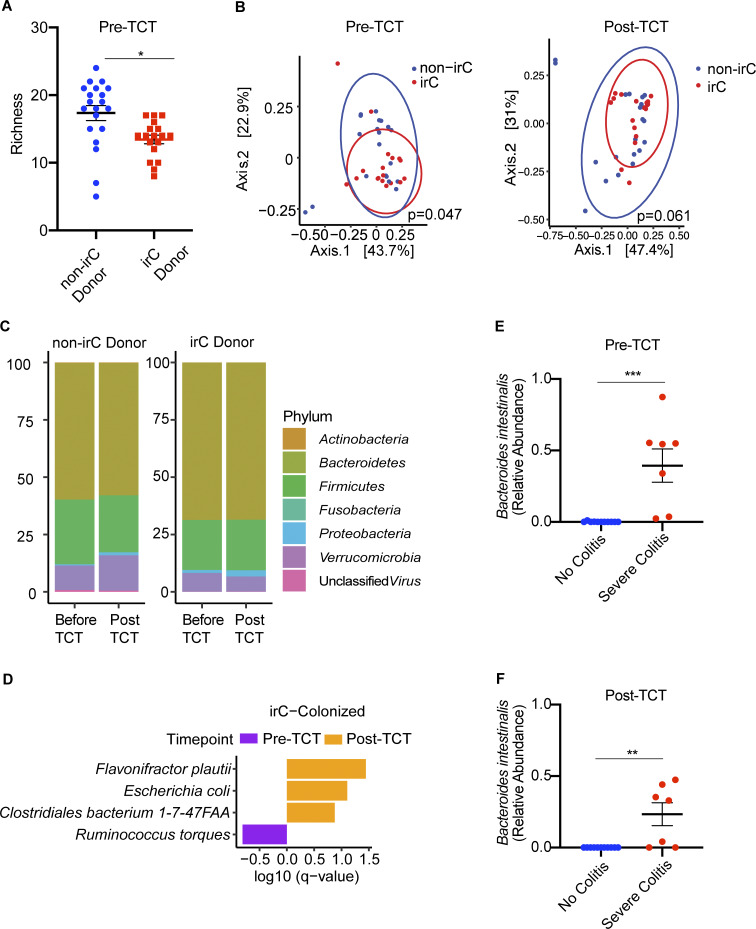
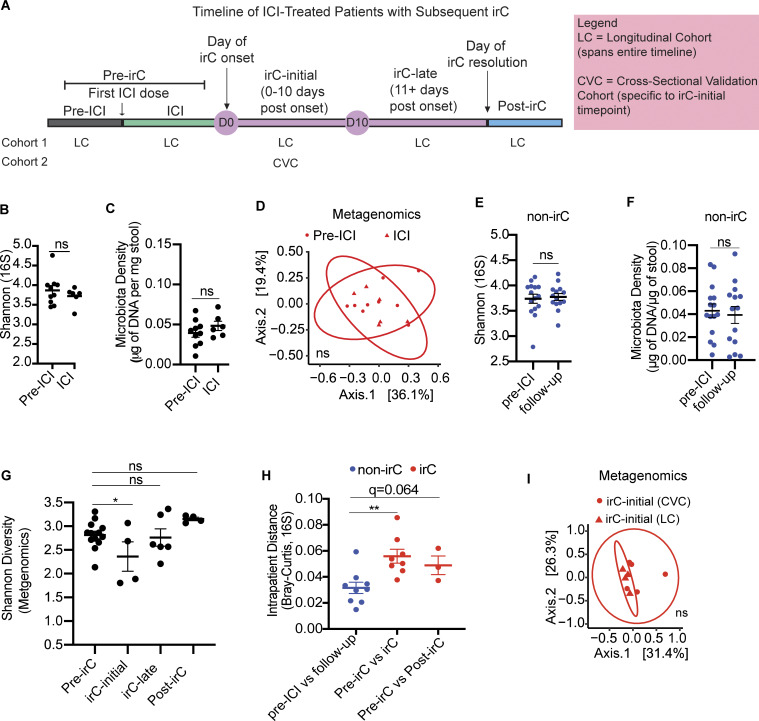
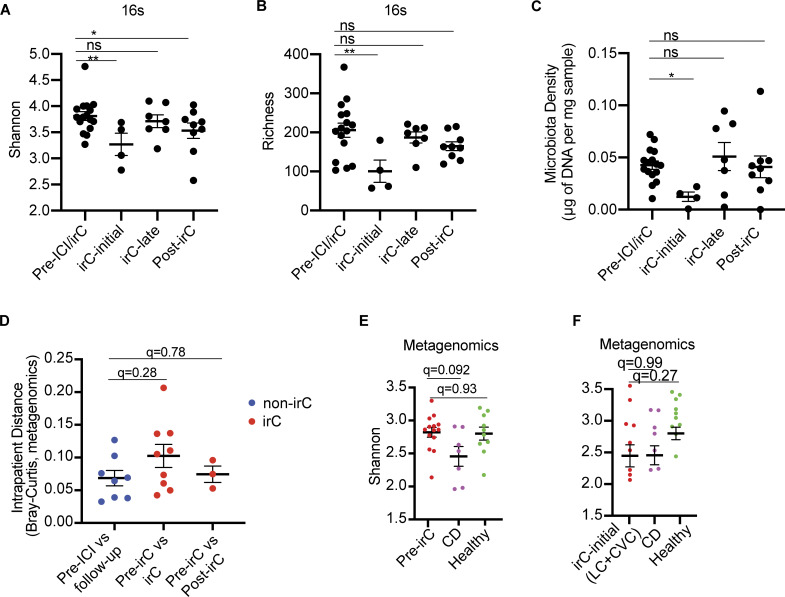
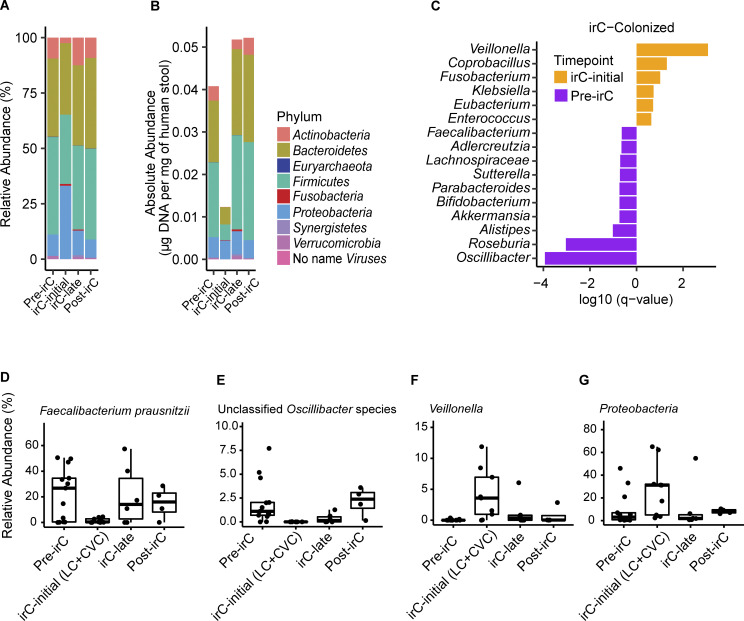
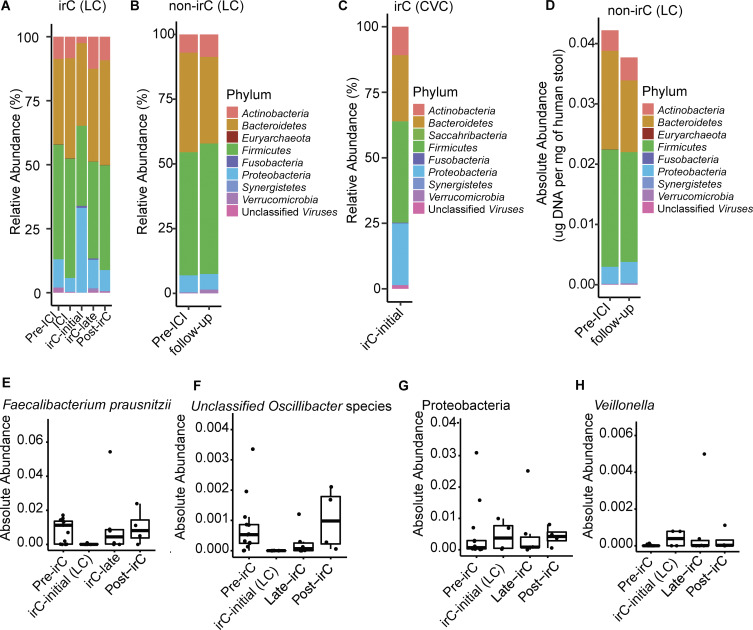
Immunotherapy-related colitis (irC) frequently emerges as an immune-related adverse event during immune checkpoint inhibitor therapy and is presumably influenced by the gut microbiota. We longitudinally studied microbiomes from 38 ICI-treated cancer patients. We compared 13 ICI-treated subjects who developed irC against 25 ICI-treated subjects who remained irC-free, along with a validation cohort. Leveraging a preclinical mouse model, predisease stools from irC subjects induced greater colitigenicity upon transfer to mice. The microbiota during the first 10 days of irC closely resembled inflammatory bowel disease microbiomes, with reduced diversity, increased Proteobacteria and Veillonella, and decreased Faecalibacterium, which normalized before irC remission. These findings highlight the irC gut microbiota as functionally distinct but phylogenetically similar to non-irC and healthy microbiomes, with the exception of an acute, transient disruption early in irC. We underscore the significance of longitudinal microbiome profiling in developing clinical avenues to detect, monitor, and mitigate irC in ICI therapy cancer patients.
© 2024 Shang et al.
Conflict of interest statement
Disclosures: R. Menon reported personal fees from Vedanta Biosciences Inc. outside the submitted work. A. Elkrief reported grants from Kanvas Bio and “other” from AstraZeneca, BMS, and Merck during the conduct of the study. J.U. Peled reported “other” from Seres Therapeutics and grants from NHLBI K08HL 143189, P30 CA008748, Society of Memorial Sloan Kettering Cancer Center, and V Foundation during the conduct of the study; personal fees from DaVolterra, CSL Behring, Crestone Inc, MaaT Pharma, Canaccord Genuity, and RA Capital and “other” from Postbiotics Plus Research and Prodigy Biosciences outside the submitted work; in addition, J.U. Peled had a patent number #15/756,845; filed 3/1/18 -METHODS AND COMPOSITIONS FOR DETECTING RISK OF CANCER RELAPSE licensed “Seres Therapeutics,” a patent number #62/977,908; filed 5/6/19 issued, and a patent number #62/843,849, filed 2/18/20 pending; and Memorial Sloan Kettering Cancer Center (MSK) has financial interests relative to Seres Therapeutics. N.J. Shah reported personal fees from Merk and grants from Exelixis and HiberCell outside the submitted work. M. Postow reported personal fees from BMS, Merck, Novartis, Eisai, Pfizer, Chugai, Erasca, Nektar, and Lyvgen, and grants from Rgenix, Infinity, BMS, Merck, Genentech, Novartis, and Bioatla outside the submitted work. J.-F. Colombel reported grants from AbbVie, Bristol Myers Squibb, and Janssen Pharmaceuticals and personal fees from Amgen, AnaptysBio, Allergan, Arena Pharmaceuticals, Astellas, Boehringer Ingelheim, Celgene Corporation, Celltrion, Eli Lilly, Ensho, Envision Pharma, Ferring Pharmaceuticals, Galmed Research, Glaxo Smith Kline, Genentech (Roche), Kaleido Biosciences, Immunic, Iterative Scopes, Merck, Landos, Microba Life Science, Novartis, Otsuka Pharmaceutical, Pfizer, Protagonist Therapeutics, Sanofi, Sun, Takeda, TiGenix, Vedanta Biosciences, Vifor, and Intestinal Biotech Development outside the submitted work. S. Gnjatic reported personal fees from Taiho Pharmaceuticals and grants from Regeneron, Bristol Myers Squibb, Takeda, Boehringer Ingelheim, Celgene, and Genentech outside the submitted work. D.M. Faleck reported personal fees from Teva, Janssen, Gilead, and Ferring outside the submitted work. J.J. Faith reported grants from Janssen Research & Development and personal fees from Vedanta Biosciences, Genfit, and Seed Health outside the submitted work. No other disclosures were reported.
Figures
References
-
- Aggarwala, V., Mogno I., Li Z., Yang C., Britton G.J., Chen-Liaw A., Mitcham J., Bongers G., Gevers D., Clemente J.C., et al. 2021. Precise quantification of bacterial strains after fecal microbiota transplantation delineates long-term engraftment and explains outcomes. Nat. Microbiol. 6:1309–1318. 10.1038/s41564-021-00966-0 - DOI - PMC - PubMed
-
- Ananthakrishnan, A.N., Luo C., Yajnik V., Khalili H., Garber J.J., Stevens B.W., Cleland T., and Xavier R.J.. 2017. Gut microbiome function predicts response to anti-integrin biologic therapy in Inflammatory Bowel diseases. Cell Host Microbe. 21:603–610.e3. 10.1016/j.chom.2017.04.010 - DOI - PMC - PubMed
-
- Andrews, M.C., Duong C.P.M., Gopalakrishnan V., Iebba V., Chen W.S., Derosa L., Khan M.A.W., Cogdill A.P., White M.G., Wong M.C., et al. 2021. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 27:1432–1441. 10.1038/s41591-021-01406-6 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
