

Genetic and functional analyses of SPTLC1 in juvenile amyotrophic lateral sclerosis
- PMID: 39666121
- PMCID: PMC11638311
- DOI: 10.1007/s00415-024-12776-5
Genetic and functional analyses of SPTLC1 in juvenile amyotrophic lateral sclerosis
Abstract
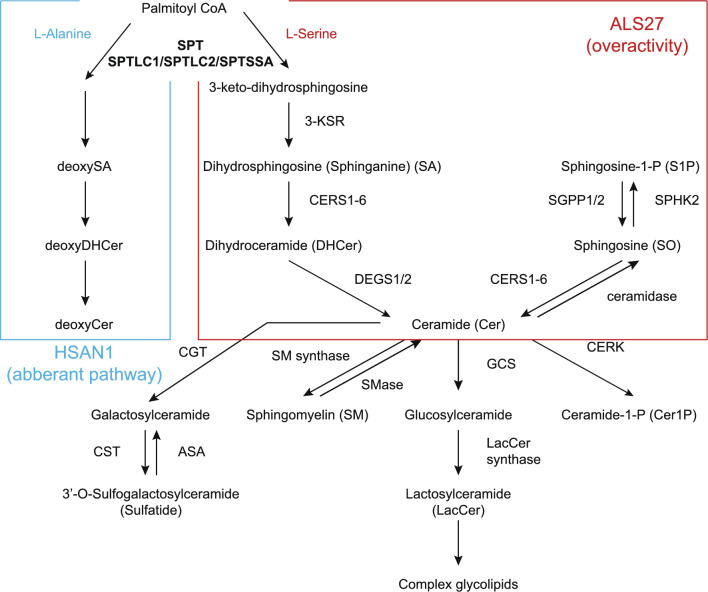
Introduction: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder of the motor system. Pathogenic variants in SPTLC1, encoding a subunit of serine palmitoyltransferase, cause hereditary sensory and autonomic neuropathy type 1 (HSAN1), and have recently been associated with juvenile ALS. SPTLC1 variants associated with ALS cause elevated levels of sphinganines and ceramides. Reports on ALS associated with SPTLC1 remain limited. This study aimed to investigate the frequency of SPTLC1 variants in ALS and relevant clinical characteristics.
Methods: We analyzed whole-exome and whole-genome sequence data from 40 probands with familial ALS and 413 patients with sporadic ALS without previously identified causative variants. Reverse transcription polymerase chain reaction (RT-PCR) analysis and droplet digital PCR (ddPCR) were used to assess splicing and mosaicism, respectively. Plasma sphingolipid levels were quantified to analyze biochemical consequences.
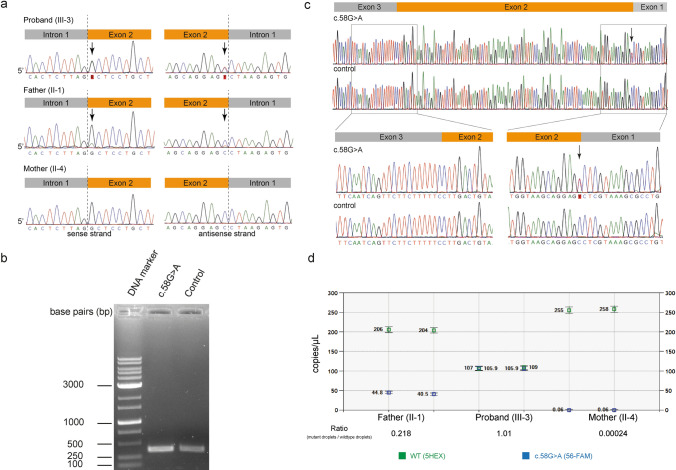
Results: The heterozygous c.58G>A, p.Ala20Thr variant was identified in a 21-year-old Japanese female patient presenting with symmetric weakness which slowly progressed over 15 years. RT-PCR analysis showed no splice defects. Plasma sphingolipid levels in the patient were significantly increased compared to her asymptomatic parents. ddPCR revealed that the asymptomatic father harbored a mosaic variant with 17% relative mutant allele abundance in peripheral blood leukocytes.
Conclusions: We identified a pathogenic c.58G>A, p.Ala20Thr SPTLC1 variant in a patient with juvenile ALS, likely inherited from an asymptomatic parent with mosaicism. Lipid analysis results are consistent with previous findings on SPTLC1-associated ALS. Further studies are necessary to determine the clinical effect of mosaic variants of SPTLC1.
Keywords: SPTLC1; Juvenile amyotrophic lateral sclerosis; Mosaicism; Sphingolipids.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Conflicts of interest: The authors declare no conflict of interest. Ethics approval: This study was approved by the Institutional Review Board of the University of Tokyo. The procedures used in this study adhere to the tenets of the Declaration of Helsinki. Consent to participate: Genomic DNA and plasma samples were obtained from all participants after obtaining written informed consent. Consent to publish: Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Figures


References
-
- Kiernan MC, Vucic S, Cheah BC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955. 10.1016/S0140-6736(10)61156-7 - PubMed
-
- Byrne S, Walsh C, Lynch C et al (2011) Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 82:623–627. 10.1136/jnnp.2010.224501 - PubMed
-
- Naruse H, Ishiura H, Mitsui J et al (2018) Molecular epidemiological study of familial amyotrophic lateral sclerosis in Japanese population by whole-exome sequencing and identification of novel HNRNPA1 mutation. Neurobiol Aging 61:255.e9-255.e16. 10.1016/j.neurobiolaging.2017.08.030 - PubMed
-
- Orban P, Devon RS, Hayden MR, Leavitt BR (2007) Chapter 15 Juvenile amyotrophic lateral sclerosis. In: Eisen AA, Shaw PJ (eds) Handbook of clinical neurology. Elsevier, Amsterdam, pp 301–312 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
