

Cellular senescence as a key contributor to secondary neurodegeneration in traumatic brain injury and stroke
- PMID: 39668354
- PMCID: PMC11636056
- DOI: 10.1186/s40035-024-00457-2
Cellular senescence as a key contributor to secondary neurodegeneration in traumatic brain injury and stroke
Abstract
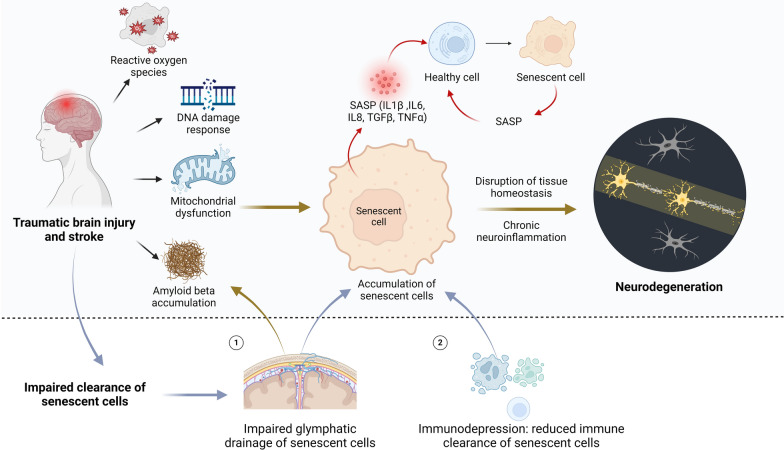
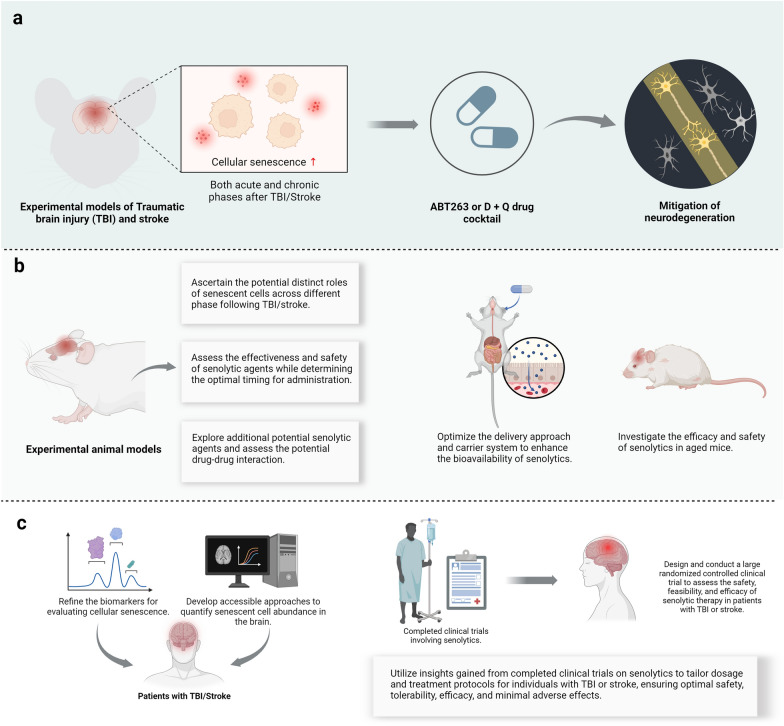
Traumatic brain injury (TBI) and stroke pose major health challenges, impacting millions of individuals globally. Once considered solely acute events, these neurological conditions are now recognized as enduring pathological processes with long-term consequences, including an increased susceptibility to neurodegeneration. However, effective strategies to counteract their devastating consequences are still lacking. Cellular senescence, marked by irreversible cell-cycle arrest, is emerging as a crucial factor in various neurodegenerative diseases. Recent research further reveals that cellular senescence may be a potential driver for secondary neurodegeneration following brain injury. Herein, we synthesize emerging evidence that TBI and stroke drive the accumulation of senescent cells in the brain. The rationale for targeting senescent cells as a therapeutic approach to combat neurodegeneration following TBI/stroke is outlined. From a translational perspective, we emphasize current knowledge and future directions of senolytic therapy for these neurological conditions.
Keywords: Cellular senescence; Neurodegeneration; Senolytic therapy; Stroke; Traumatic brain injury.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: All authors have read and approved the final manuscript for publication. Competing interests: The authors declare that they have no competing interests.
Figures


References
-
- Capizzi A, Woo J, Verduzco-Gutierrez M. Traumatic brain injury: an overview of epidemiology, pathophysiology, and medical management. Med Clin North Am. 2020;104(2):213–38. - PubMed
-
- Saini V, Guada L, Yavagal DR. Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology. 2021;97(20 Suppl 2):S6–16. - PubMed
-
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24(2):133–50. - PubMed
-
- Kunz A, Dirnagl U, Mergenthaler P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract Res Clin Anaesthesiol. 2010;24(4):495–509. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
