

This is a preprint.
Identification of potential riboswitch elements in Homo Sapiens mRNA 5'UTR sequences using Positive-Unlabeled machine learning
- PMID: 39677788
- PMCID: PMC11642740
- DOI: 10.1101/2023.11.23.568398
Identification of potential riboswitch elements in Homo Sapiens mRNA 5'UTR sequences using Positive-Unlabeled machine learning
Update in
-
Identification of potential riboswitch elements in Homo sapiens mRNA 5'UTR sequences using positive-unlabeled machine learning.PLoS One. 2025 Apr 24;20(4):e0320282. doi: 10.1371/journal.pone.0320282. eCollection 2025. PLoS One. 2025. PMID: 40273288 Free PMC article.
Abstract
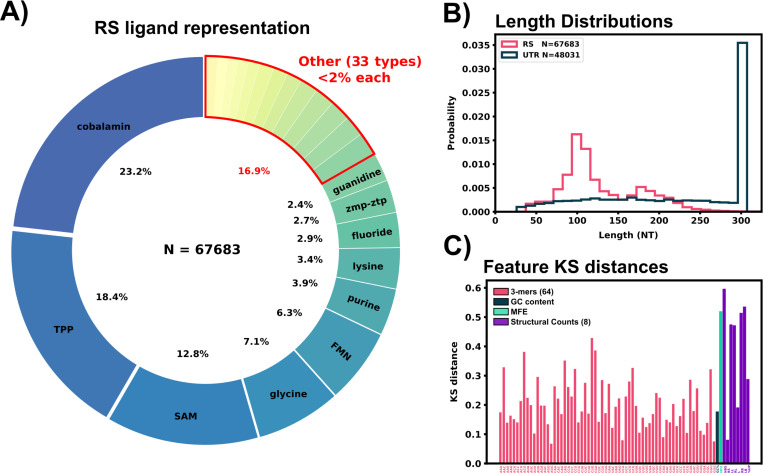
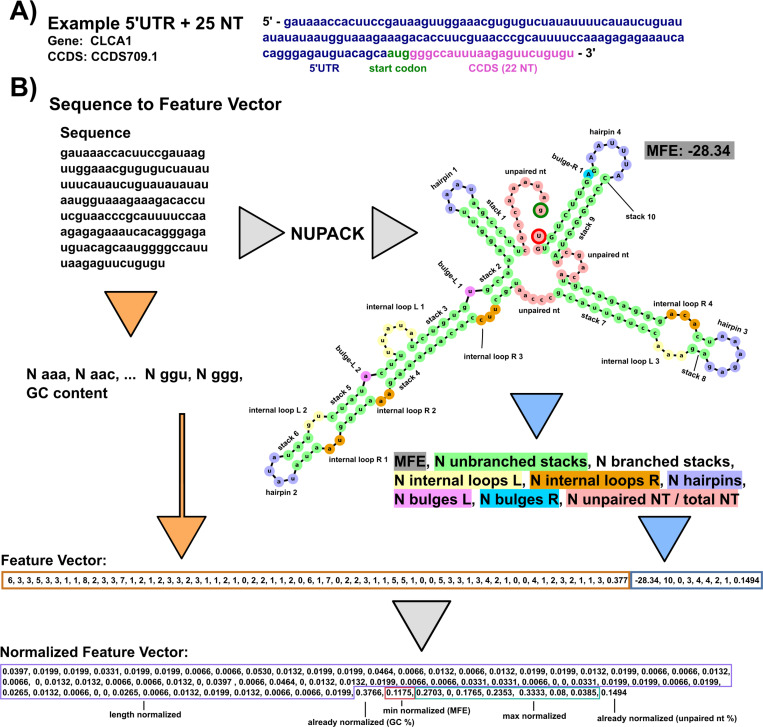
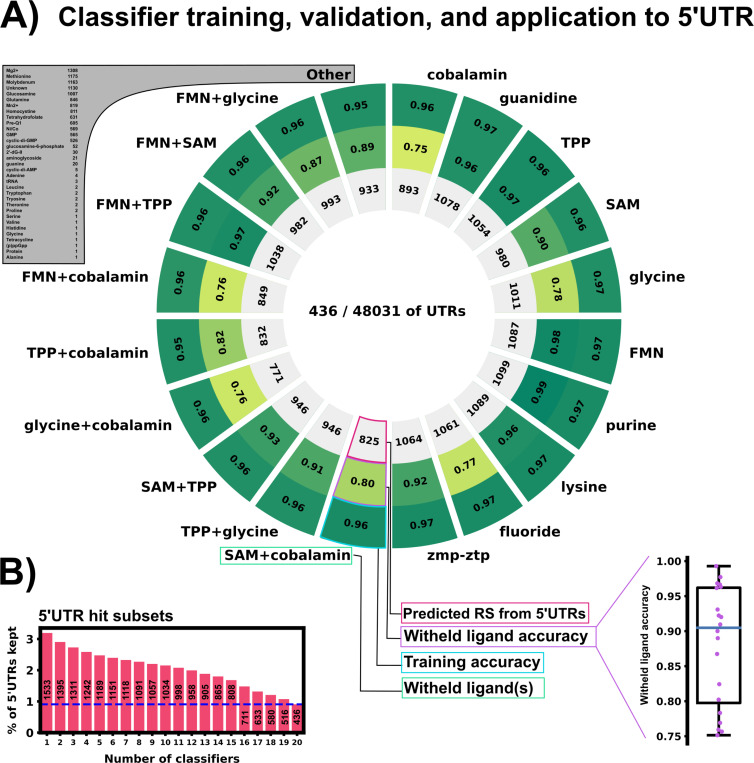
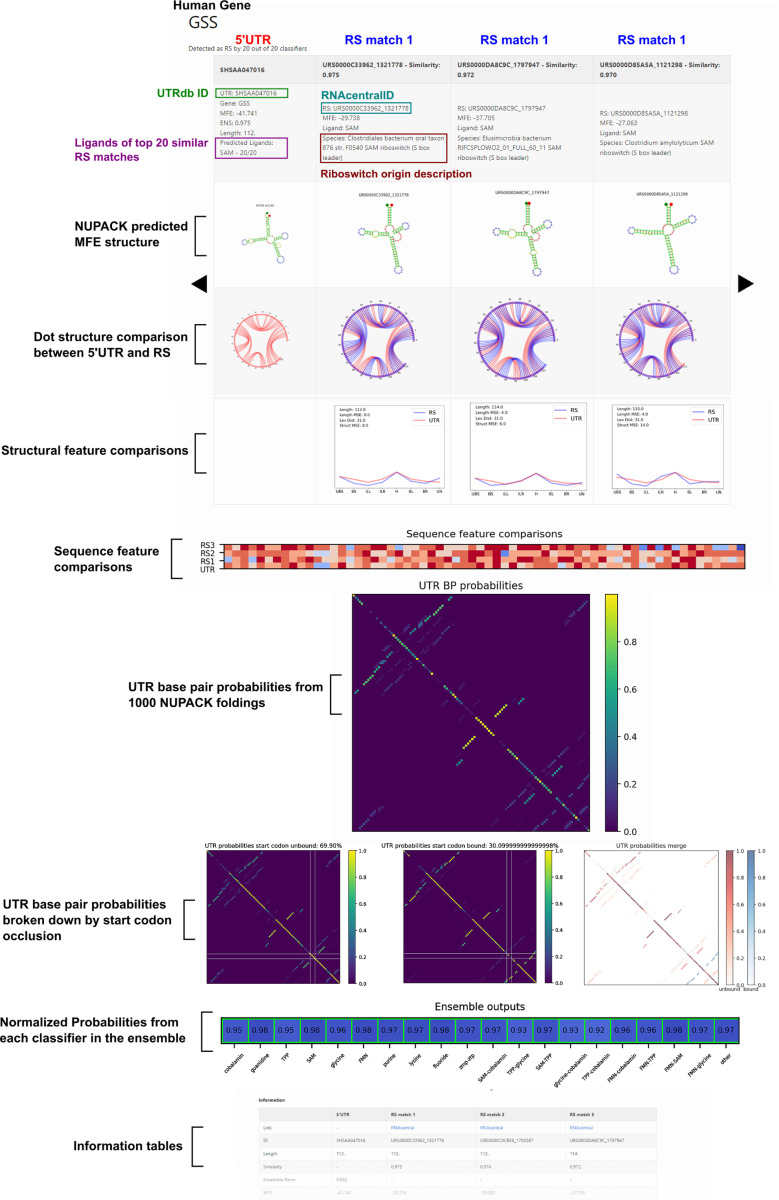
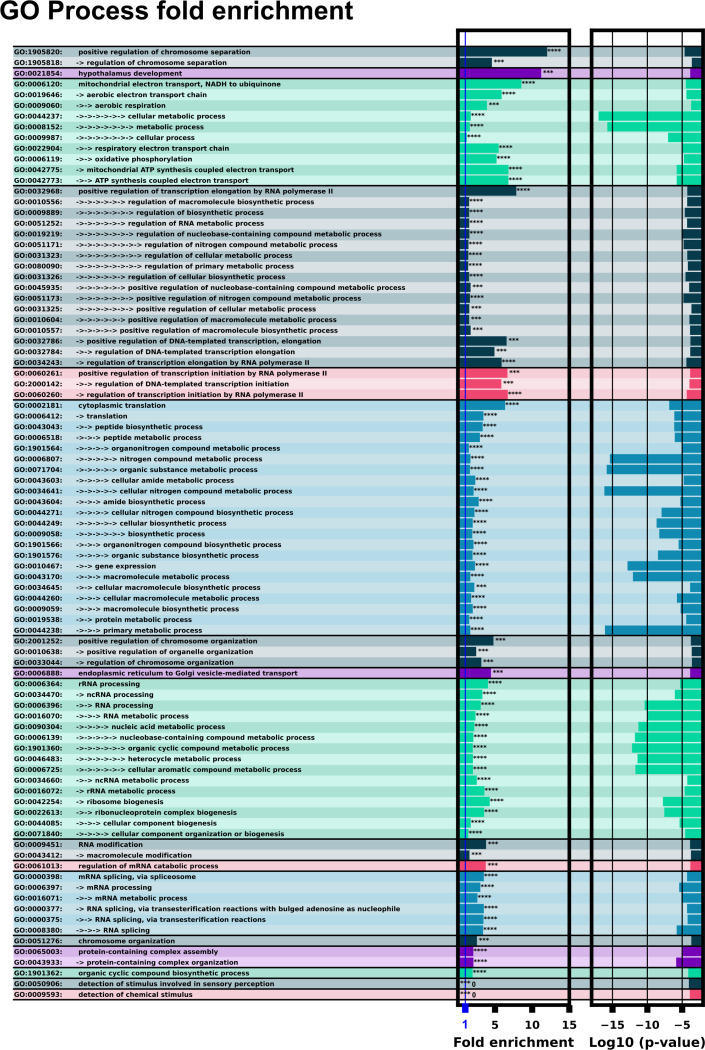
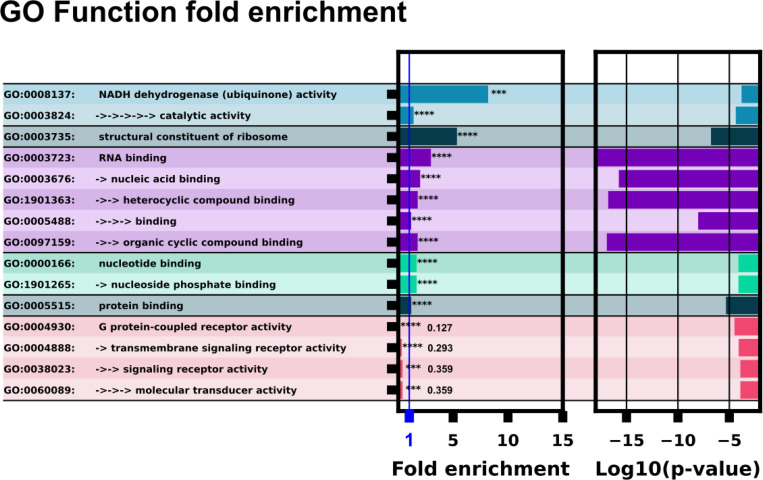
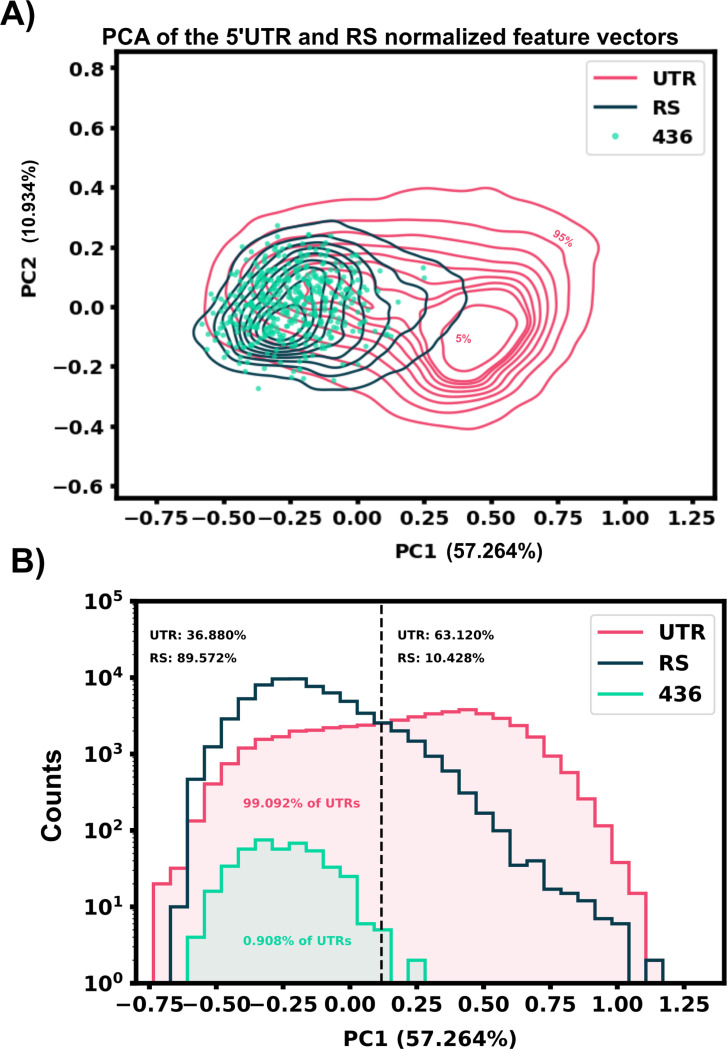
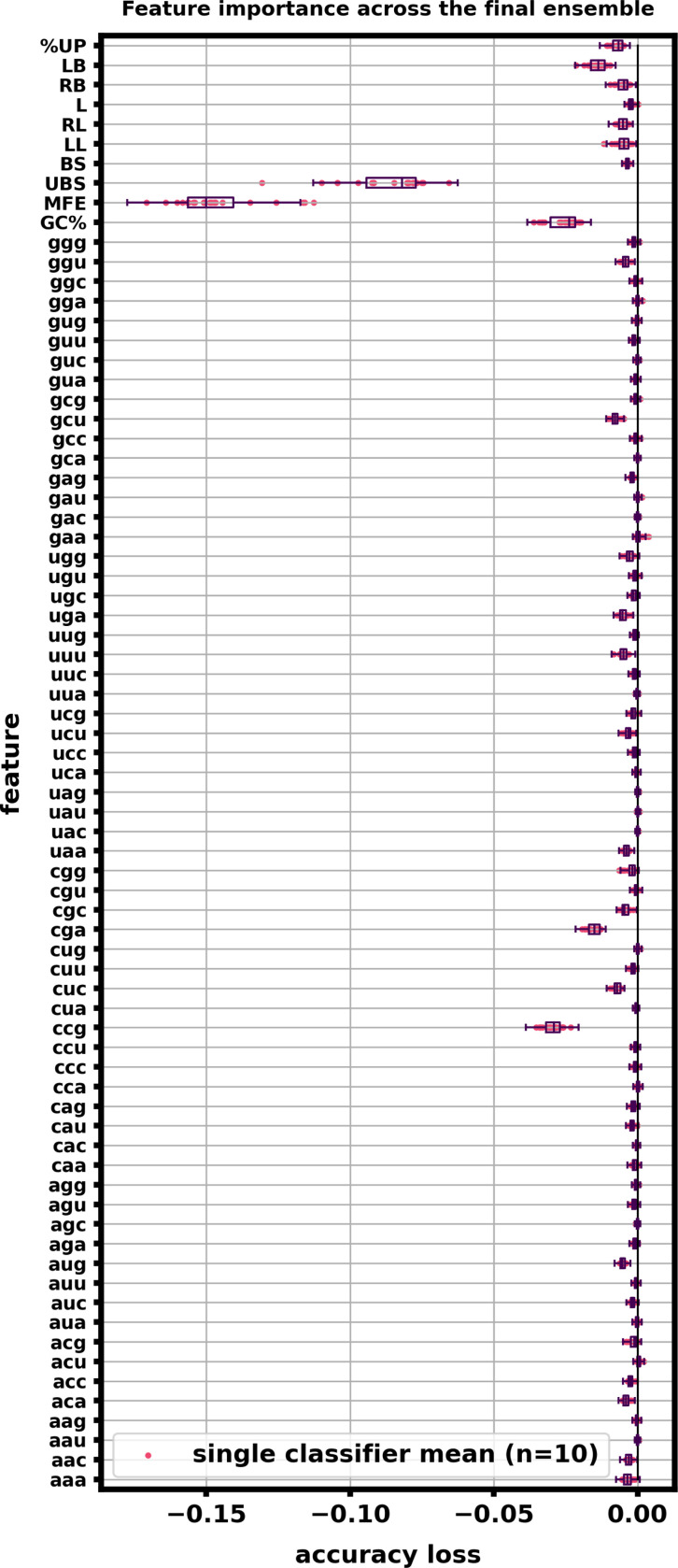
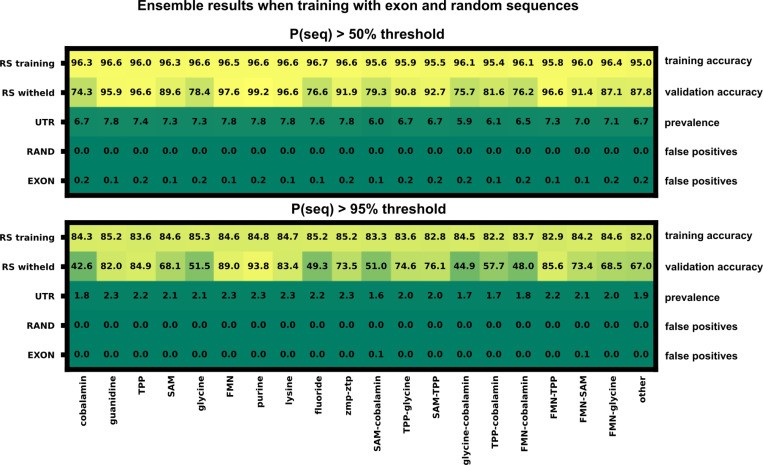
Riboswitches are a class of noncoding RNA structures that interact with target ligands to cause a conformational change that can then execute some regulatory purpose within the cell. Riboswitches are ubiquitous and well characterized in bacteria and prokaryotes, with additional examples also being found in fungi, plants, and yeast. To date, no purely RNA-small molecule riboswitch has been discovered in Homo Sapiens. Several analogous riboswitch-like mechanisms have been described within the H. Sapiens translatome within the past decade, prompting the question: Is there a H. Sapiens riboswitch dependent on only small molecule ligands? In this work, we set out to train positive unlabeled machine learning classifiers on known riboswitch sequences and apply the classifiers to H. Sapiens mRNA 5'UTR sequences found in the 5'UTR database, UTRdb, in the hope of identifying a set of mRNAs to investigate for riboswitch functionality. 67,683 riboswitch sequences were obtained from RNAcentral and sorted for ligand type and used as positive examples and 48,031 5'UTR sequences were used as unlabeled, unknown examples. Positive examples were sorted by ligand, and 20 positive-unlabeled classifiers were trained on sequence and secondary structure features while withholding one or two ligand classes. Cross validation was then performed on the withheld ligand sets to obtain a validation accuracy range of 75%-99%. The joint sets of 5'UTRs identified as potential riboswitches by the 20 classifiers were then analyzed. 15333 sequences were identified as a riboswitch by one or more classifier(s) and 436 of the H. Sapiens 5'UTRs were labeled as harboring potential riboswitch elements by all 20 classifiers. These 436 sequences were mapped back to the most similar riboswitches within the positive data and examined. An online database of identified and ranked 5'UTRs, their features, and their most similar matches to known riboswitches, is provided to guide future experimental efforts to identify H. Sapiens riboswitches.
Conflict of interest statement
Conflict of Interest Statement The authors declare the absence of any commercial or financial relationships that could be construed as a conflict of interest for this research.
Figures


Similar articles
-
Identification of potential riboswitch elements in Homo sapiens mRNA 5'UTR sequences using positive-unlabeled machine learning.PLoS One. 2025 Apr 24;20(4):e0320282. doi: 10.1371/journal.pone.0320282. eCollection 2025. PLoS One. 2025. PMID: 40273288 Free PMC article.
-
Riboswitch detection using profile hidden Markov models.BMC Bioinformatics. 2009 Oct 8;10:325. doi: 10.1186/1471-2105-10-325. BMC Bioinformatics. 2009. PMID: 19814811 Free PMC article.
-
The dynamic nature of RNA as key to understanding riboswitch mechanisms.Acc Chem Res. 2011 Dec 20;44(12):1339-48. doi: 10.1021/ar200035g. Epub 2011 Jun 16. Acc Chem Res. 2011. PMID: 21678902
-
Computational prediction of riboswitches.Methods Enzymol. 2015;553:287-312. doi: 10.1016/bs.mie.2014.10.063. Epub 2015 Feb 19. Methods Enzymol. 2015. PMID: 25726470 Review.
-
Machine Learning for Designing Next-Generation mRNA Therapeutics.Acc Chem Res. 2022 Jan 4;55(1):24-34. doi: 10.1021/acs.accounts.1c00621. Epub 2021 Dec 14. Acc Chem Res. 2022. PMID: 34905691 Review.
References
-
- Abreu-Goodger C. and Merino E.. RibEx: a web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic Acids Research, 33:W690, July 2005. URL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1160206/, doi:10.1093/NAR/GKI445. - DOI - PMC - PubMed
-
- Amin N., McGrath A., and Chen Y. P. P.. Evaluation of deep learning in non-coding RNA classification. Nature Machine Intelligence, 1:246–256, May 2019. URL: https://www.nature.com/articles/s42256-019-0051-2, doi:10.1038/s42256-019-0051-2. - DOI
-
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., and Sherlock G.. Gene Ontology: tool for the unification of biology NIH public access author manuscript. Nature Genetics, 25:25–29, May 2000. doi:10.1038/75556. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous