

This is a preprint.
CGC1, a new reference genome for Caenorhabditis elegans
- PMID: 39677790
- PMCID: PMC11643116
- DOI: 10.1101/2024.12.04.626850
CGC1, a new reference genome for Caenorhabditis elegans
Update in
-
CGC1, a new reference genome for Caenorhabditis elegans.Genome Res. 2025 Aug 1;35(8):1902-1918. doi: 10.1101/gr.280274.124. Genome Res. 2025. PMID: 40664475 Free PMC article.
Abstract
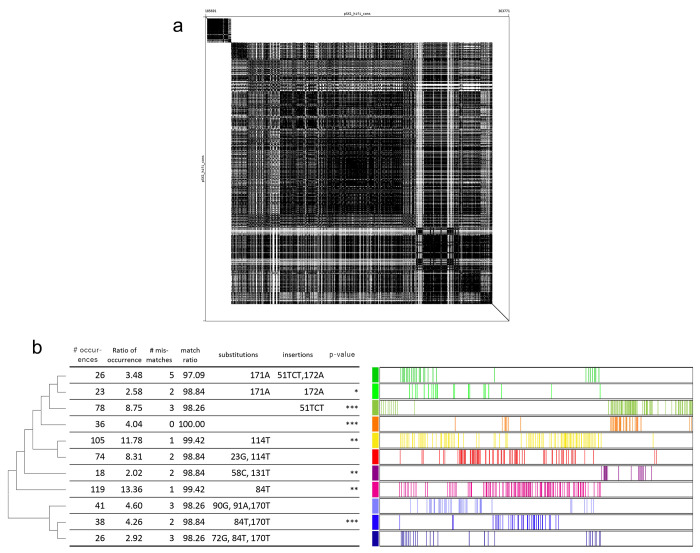
The original 100.3 Mb reference genome for Caenorhabditis elegans, generated from the wild-type laboratory strain N2, has been crucial for analysis of C. elegans since 1998 and has been considered complete since 2005. Unexpectedly, this long-standing reference was shown to be incomplete in 2019 by a genome assembly from the N2-derived strain VC2010. Moreover, genetically divergent versions of N2 have arisen over decades of research and hindered reproducibility of C. elegans genetics and genomics. Here we provide a 106.4 Mb gap-free, telomere-to-telomere genome assembly of C. elegans, generated from CGC1, an isogenic derivative of the N2 strain. We used improved long-read sequencing and manual assembly of 43 recalcitrant genomic regions to overcome deficiencies of prior N2 and VC2010 assemblies, and to assemble tandem repeat loci including a 772-kb sequence for the 45S rRNA genes. While many differences from earlier assemblies came from repeat regions, unique additions to the genome were also found. Of 19,972 protein-coding genes in the N2 assembly, 19,790 (99.1%) encode products that are unchanged in the CGC1 assembly. The CGC1 assembly also may encode 183 new protein-coding and 163 new ncRNA genes. CGC1 thus provides both a completely defined reference genome and corresponding isogenic wild-type strain for C. elegans, allowing unique opportunities for model and systems biology.
Figures





References
-
- Boeke J. D., Church G., Hessel A., Kelley N. J., Arkin A. et al. 2016. Genome engineering: the Genome Project-Write. Science 353: 126–127. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous