

This is a preprint.
A robust benchmark for detecting low-frequency variants in the HG002 Genome In A Bottle NIST reference material
- PMID: 39677813
- PMCID: PMC11642750
- DOI: 10.1101/2024.12.02.625685
A robust benchmark for detecting low-frequency variants in the HG002 Genome In A Bottle NIST reference material
Abstract
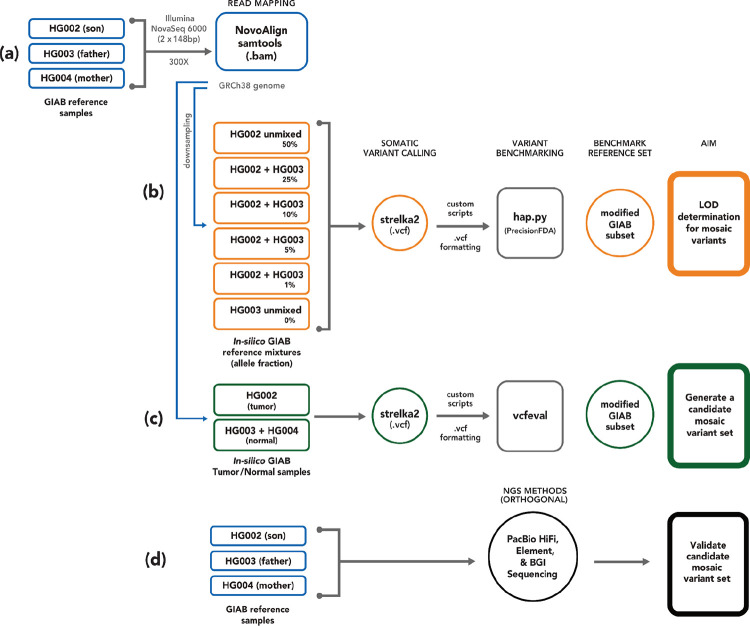
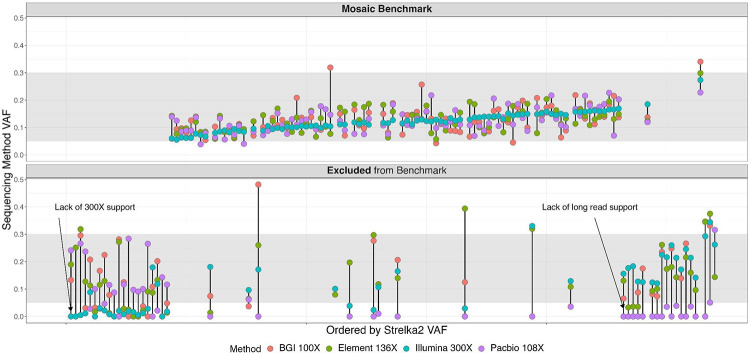
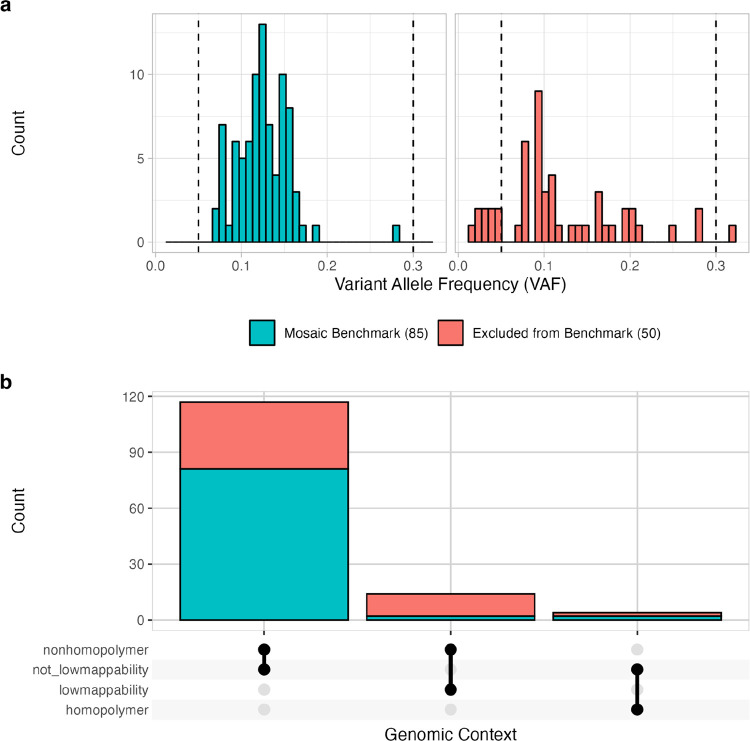
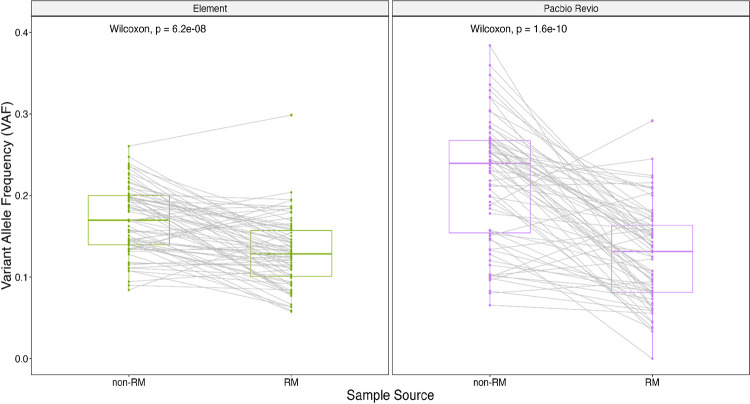
Somatic mosaicism is an important cause of disease, but mosaic and somatic variants are often challenging to detect because they exist in only a fraction of cells. To address the need for benchmarking subclonal variants in normal cell populations, we developed a benchmark containing mosaic variants in the Genome in a Bottle Consortium (GIAB) HG002 reference material DNA from a large batch of a normal lymphoblastoid cell line. First, we used a somatic variant caller with high coverage (300x) Illumina whole genome sequencing data from the Ashkenazi Jewish trio to detect variants in HG002 not detected in at least 5% of cells from the combined parental data. These candidate mosaic variants were subsequently evaluated using >100x BGI, Element, and PacBio HiFi data. High confidence candidate SNVs with variant allele fractions above 5% were included in the HG002 draft mosaic variant benchmark, with 13/85 occurring in medically relevant gene regions. We also delineated a 2.45 Gbp subset of the previously defined germline autosomal benchmark regions for HG002 in which no additional mosaic variants >2% exist, enabling robust assessment of false positives. The variant allele fraction of some mosaic variants is different between batches of cells, so using data from the homogeneous batch of reference material DNA is critical for benchmarking these variants. External validation of this mosaic benchmark showed it can be used to reliably identify both false negatives and false positives for a variety of technologies and detection algorithms, demonstrating its utility for optimization and validation. By adding our characterization of mosaic variants in this widely-used cell line, we support extensive benchmarking efforts using it in simulation, spike-in, and mixture studies.
Keywords: SNV; Somatic mosaicism; benchmarking; genome in a bottle; genome sequencing; mosaic variant; somatic variant; variant calling.
Conflict of interest statement
Competing Interests C.E.M. is a co-Founder of Onegevity. Y.W., M.R., A.V., L.M., W.C., S.C., J.H., R.M., and G.P. are Illumina employees and equity owners. A.C., P.C., K.S., D.C., A.K., and L.B. are employees of Google LLC and receive equity compensation. P.C.B. sits on the scientific advisory boards of Intersect Diagnostics Inc. and BioSymetrics Inc., and previously sat on that of Sage Bionetworks.
Figures
References
-
- Agresti Alan, and Coull Brent A.. 1998. “Approximate Is Better than ‘Exact’ for Interval Estimation of Binomial Proportions.” The American Statistician 52 (2): 119–26.
-
- Ball Madeleine P., Thakuria Joseph V., Alexander Wait Zaranek Tom Clegg, Rosenbaum Abraham M., Wu Xiaodi, Angrist Misha, et al. 2012. “A Public Resource Facilitating Clinical Use of Genomes.” Proceedings of the National Academy of Sciences of the United States of America 109 (30): 11920–27. - PMC - PubMed
-
- Brown Lawrence D., Cai T. Tony, and DasGupta Anirban. 2001. “Interval Estimation for a Binomial Proportion.” Schweizerische Monatsschrift Fur Zahnheilkunde = Revue Mensuelle Suisse D’odonto-Stomatologie / SSO 16 (2): 101–33.
-
- Chen Lixin, Liu Pingfang, Evans Thomas C. Jr, and Ettwiller Laurence M.. 2017. “DNA Damage Is a Pervasive Cause of Sequencing Errors, Directly Confounding Variant Identification.” Science 355 (6326): 752–56. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials