

epiTCR-KDA: knowledge distillation model on dihedral angles for TCR-peptide prediction
- PMID: 39678207
- PMCID: PMC11646569
- DOI: 10.1093/bioadv/vbae190
epiTCR-KDA: knowledge distillation model on dihedral angles for TCR-peptide prediction
Abstract
Motivation: The prediction of the T-cell receptor (TCR) and antigen bindings is crucial for advancements in immunotherapy. However, most current TCR-peptide interaction predictors struggle to perform well on unseen data. This limitation may stem from the conventional use of TCR and/or peptide sequences as input, which may not adequately capture their structural characteristics. Therefore, incorporating the structural information of TCRs and peptides into the prediction model is necessary to improve its generalizability.
Results: We developed epiTCR-KDA (KDA stands for Knowledge Distillation model on Dihedral Angles), a new predictor of TCR-peptide binding that utilizes the dihedral angles between the residues of the peptide and the TCR as a structural descriptor. This structural information was integrated into a knowledge distillation model to enhance its generalizability. epiTCR-KDA demonstrated competitive prediction performance, with an area under the curve (AUC) of 1.00 for seen data and AUC of 0.91 for unseen data. On public datasets, epiTCR-KDA consistently outperformed other predictors, maintaining a median AUC of 0.93. Further analysis of epiTCR-KDA revealed that the cosine similarity of the dihedral angle vectors between the unseen testing data and training data is crucial for its stable performance. In conclusion, our epiTCR-KDA model represents a significant step forward in developing a highly effective pipeline for antigen-based immunotherapy.
Availability and implementation: epiTCR-KDA is available on GitHub (https://github.com/ddiem-ri-4D/epiTCR-KDA).
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
The authors have no conflicts of interest to declare, except that M.-D.P. holds shares in NexCalibur Therapeutics, the company that has provided funding for the research presented in this publication.
Figures
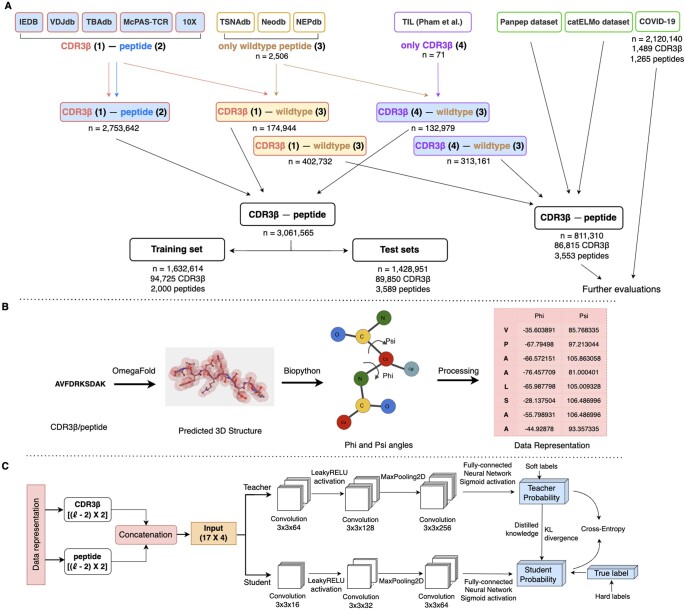
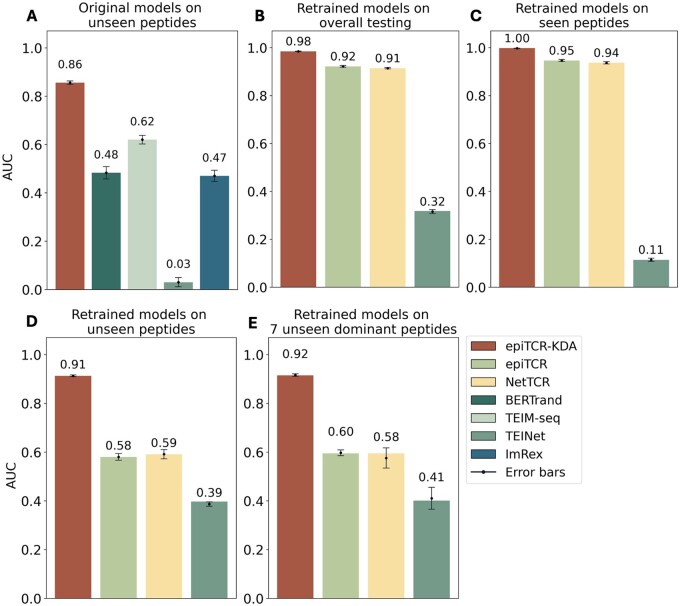
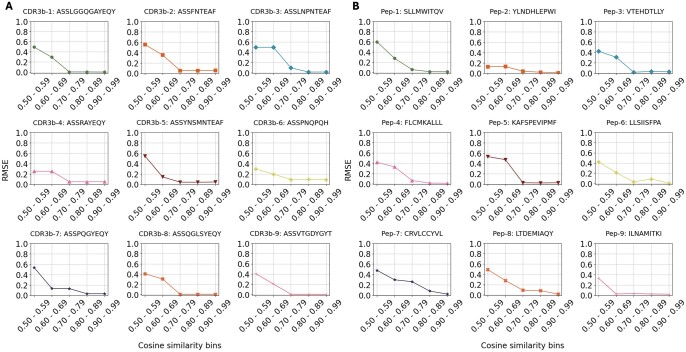
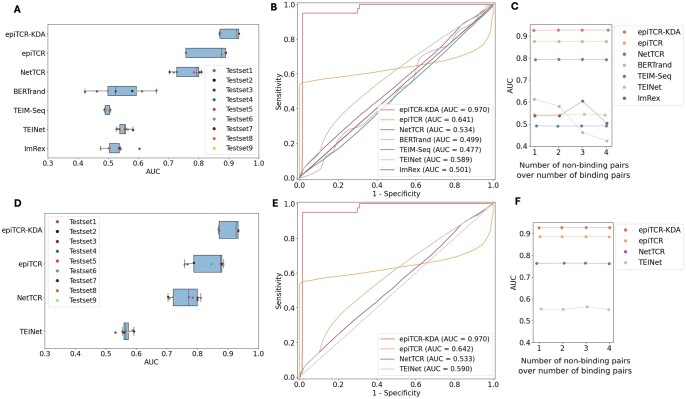
References
-
- A New Way of Exploring Immunity—Linking Highly Multiplexed Antigen Recognition to Immune Repertoire and Phenotype | Technology Networks. (2020). Retrieved March 22, 2024. https://www.technologynetworks.com/immunology/application-notes/a-new-wa...
-
- Bio.PDB.internal_coords module–Biopython 1.84.dev0 documentation. (n.d.). Retrieved March 22, 2024. https://biopython.org/docs/dev/api/Bio.PDB.internal_coords.html
-
- Chawla A, Yin H, Molchanov P. et al. Data-free knowledge distillation for object detection. Proceedings - 2021 IEEE Winter Conference on Applications of Computer Vision, WACV 2021, p.3288–97, 2021. 10.1109/WACV48630.2021.00333 - DOI
-
- Croce G, Lani R, Tardivon D. et al. Phage display profiling of CDR3β loops enables machine learning predictions of NY-ESO-1 specific TCRs. BioRxiv, 2024, 10.1101/2024.06.27.600973, preprint: not peer reviewed. - DOI
LinkOut - more resources
Full Text Sources
