

Susceptibility of Mitophagy-Deficient Tumors to Ferroptosis Induction by Relieving the Suppression of Lipid Peroxidation
- PMID: 39679775
- PMCID: PMC11809388
- DOI: 10.1002/advs.202412593
Susceptibility of Mitophagy-Deficient Tumors to Ferroptosis Induction by Relieving the Suppression of Lipid Peroxidation
Abstract
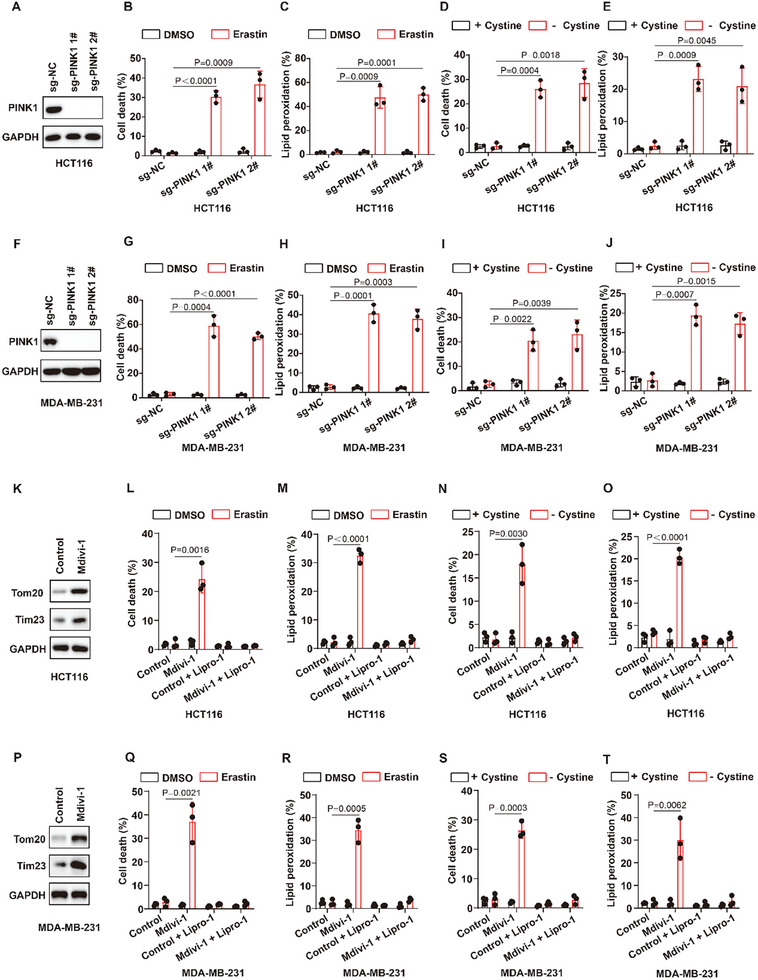
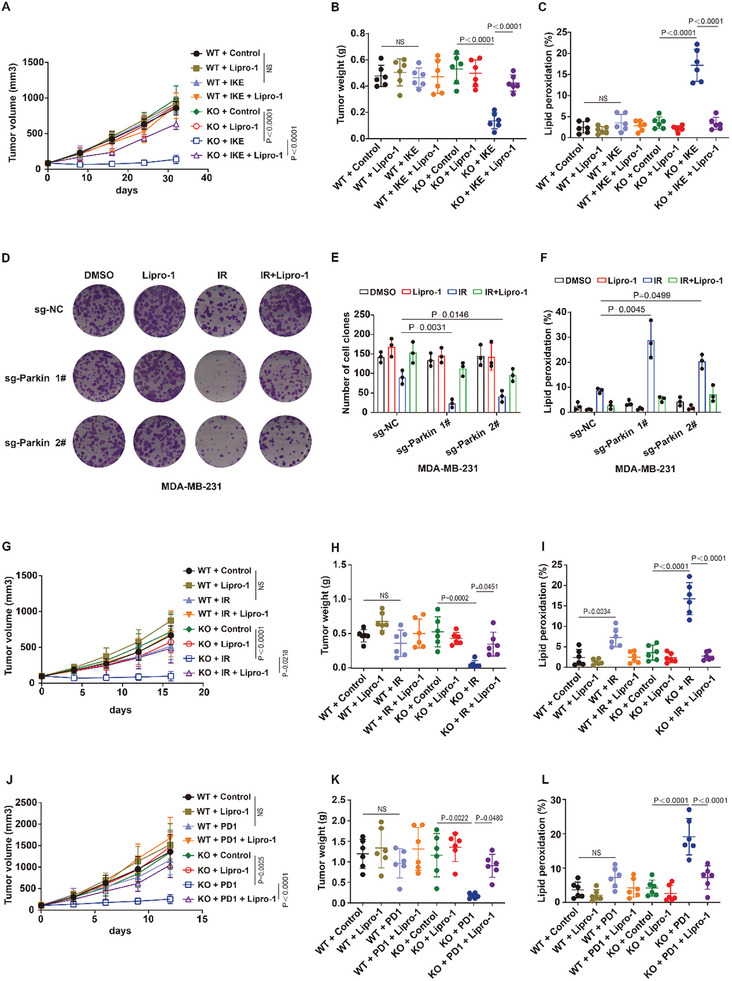
The identification of ferroptosis-sensitive cancers is critical for the application of ferroptosis-inducing therapies in cancer therapy. Here, patient-derived organoid screening models of colorectal cancer are established to identify tumors that are sensitive to ferroptosis-inducing therapy. This study discovers that patient-derived tumors characterized by mitophagy deficiency are hypersensitive to ferroptosis-inducing therapies. Mechanistically, a novel negative feedback regulatory pathway of lipid peroxidation is identified, which is one of the important intrinsic anti-ferroptosis mechanisms of cancer cells. Lipid peroxidation-mediated endoplasmic reticulum stress transcriptionally upregulates Parkin to promote mitophagy through ATF4. Mitophagy limits the generation of lipid peroxidation products and subsequently inhibits ferroptosis by inhibiting the accumulation of mitochondrial ROS. Mitophagy-deficient tumors lack this anti-ferroptotic mechanism, unleashing the generation of lipid peroxidation and potent ferroptotic cell death induced by erastin, RSL3, cysteine deprivation, radiotherapy, and immunotherapy. More importantly, ferroptosis-inducing therapy selectively inhibits the growth and distant metastasis of mitophagy-deficient tumors in vivo. In summary, patient-derived organoids of colorectal cancer patients for screening ferroptosis-sensitive tumors are established, providing a paradigm for identifying that patient-derived tumors are sensitive to ferroptosis-inducing therapies. This study concludes that mitophagy-deficient tumors are vulnerable to ferroptosis induction, which may lead to the development of new therapeutic strategies for tumors deficient in mitophagy.
Keywords: ferroptosis; lipid peroxidation; mitophagy‐deficient tumors; parkin.
© 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Zhang H. L., Hu B. X., Li Z. L., Du T., Shan J. L., Ye Z. P., Peng X. D., Li X., Huang Y., Zhu X. Y., Chen Y. H., Feng G. K., Yang D., Deng R., Zhu X. F., Nat. Cell Biol. 2022, 24, 88. - PubMed
-
- Zhang H. L., Hu B. X., Ye Z. P., Li Z. L., Liu S., Zhong W. Q., Du T., Yang D., Mai J., Li L. C., Chen Y. H., Zhu X. Y., Li X., Feng G. K., Zhu X. F., Deng R., Sci. Transl. Med. 2024, 16, eadk0330. - PubMed
-
- Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernandez‐Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R., Rata M., Koh D. M., Tunariu N., Collins D., Hulkki‐Wilson S., Ragulan C., Spiteri I., Moorcraft S. Y., Chau I., Rao S., Watkins D., Fotiadis N., Bali M., Darvish‐Damavandi M., Lote H., Eltahir Z., Smyth E. C., Begum R., Clarke P. A., Hahne J. C., et al., Science 2018, 359, 920. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical