

A common form of dominant human IFNAR1 deficiency impairs IFN-α and -ω but not IFN-β-dependent immunity
- PMID: 39680367
- PMCID: PMC11648951
- DOI: 10.1084/jem.20241413
A common form of dominant human IFNAR1 deficiency impairs IFN-α and -ω but not IFN-β-dependent immunity
Abstract
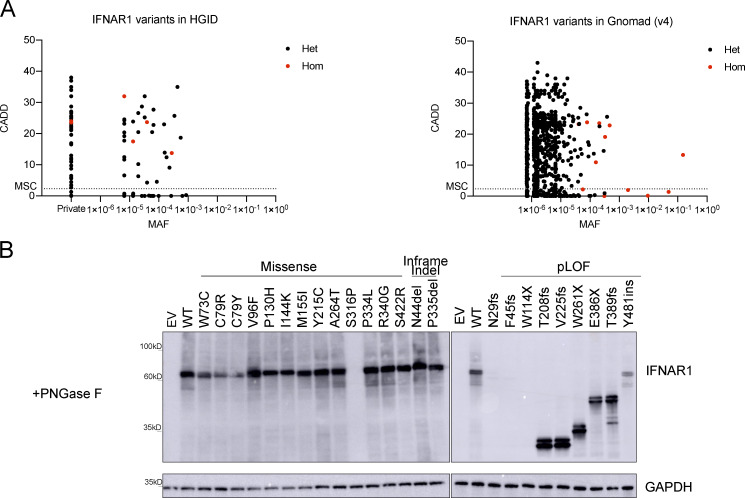
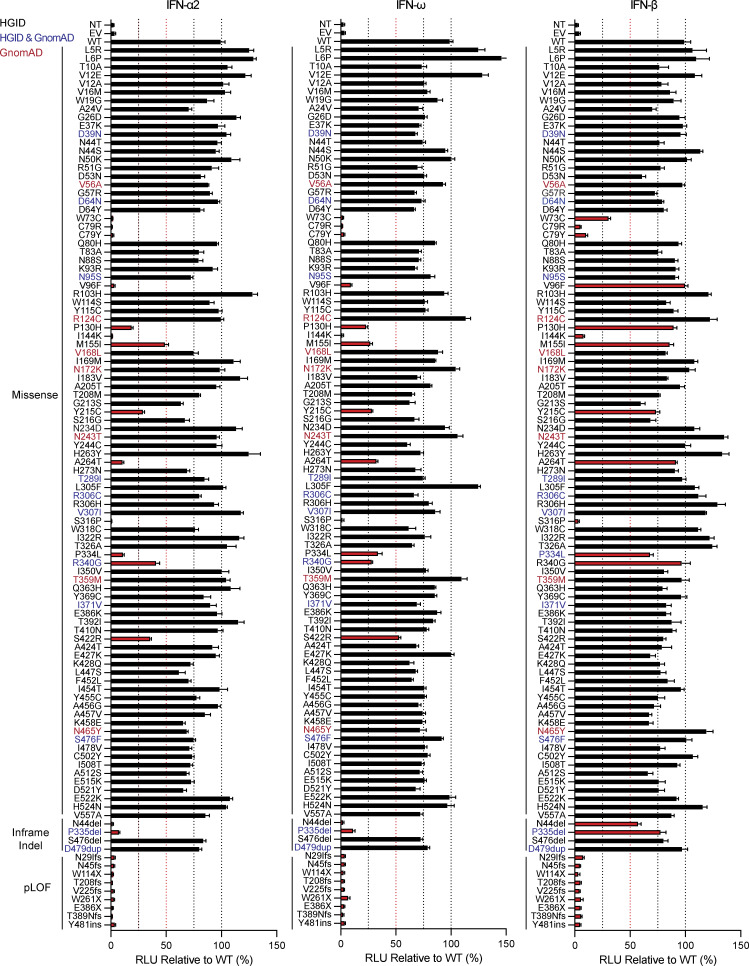
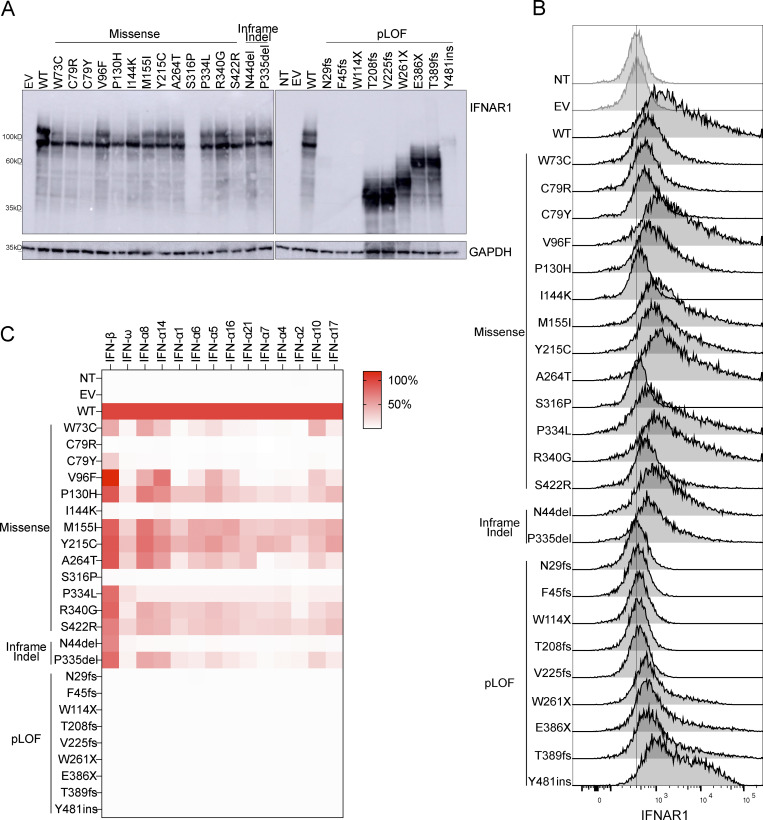
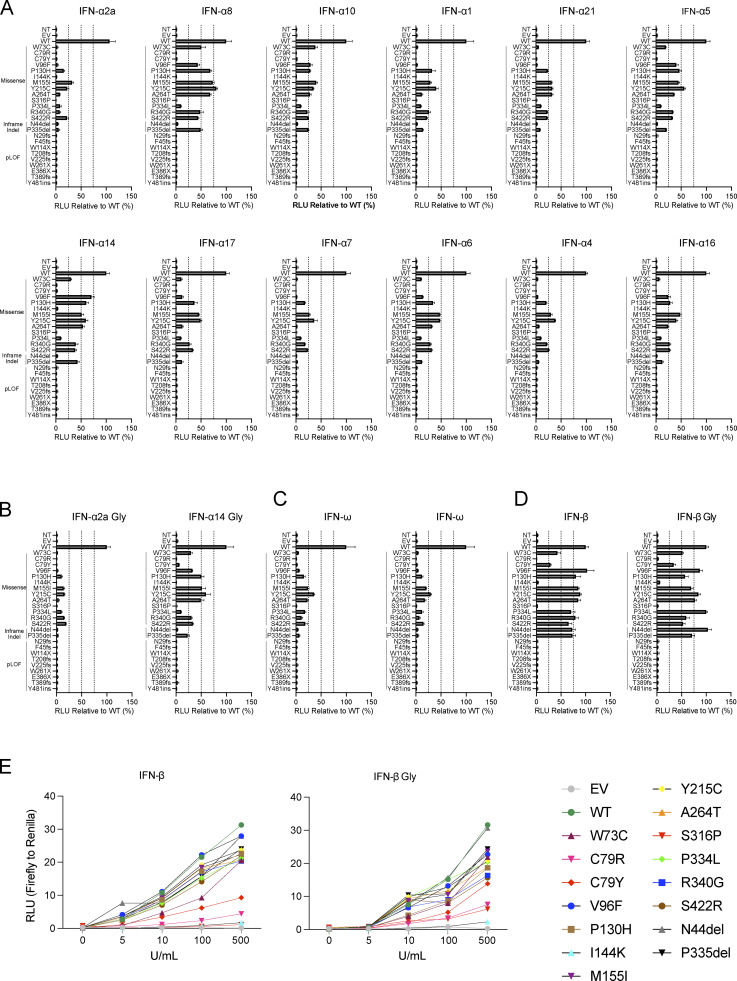
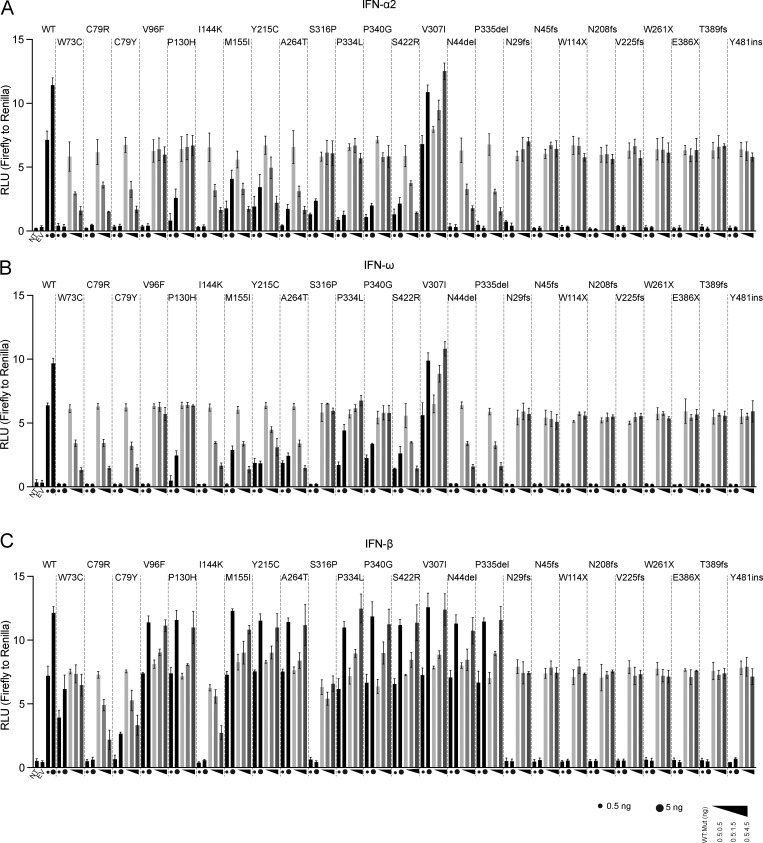
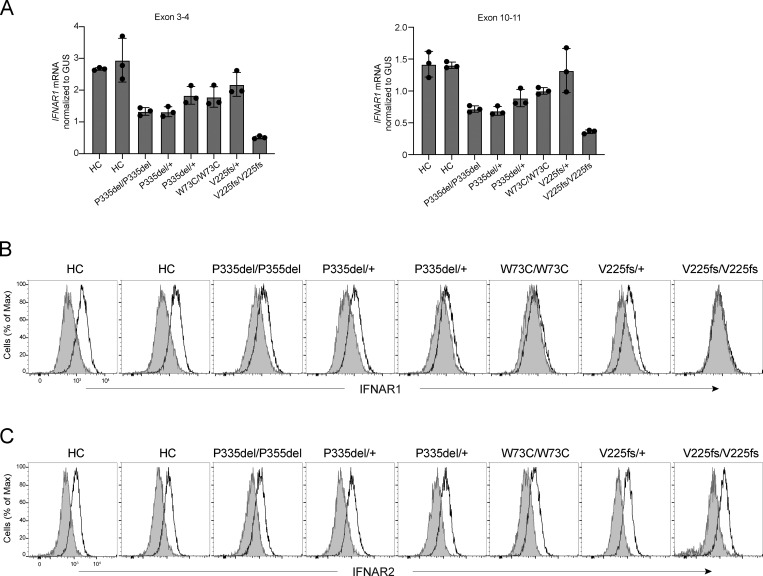
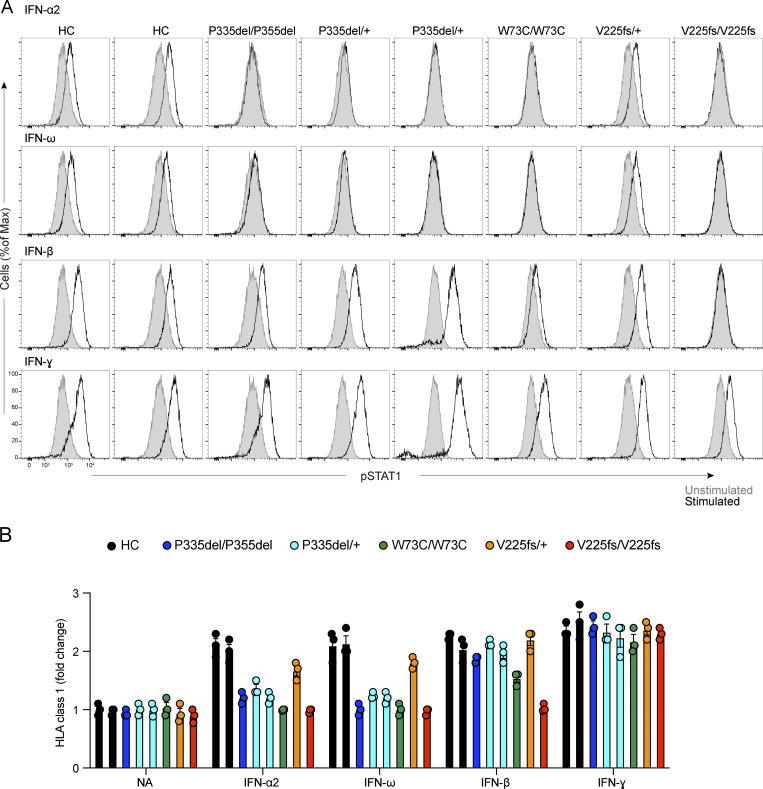
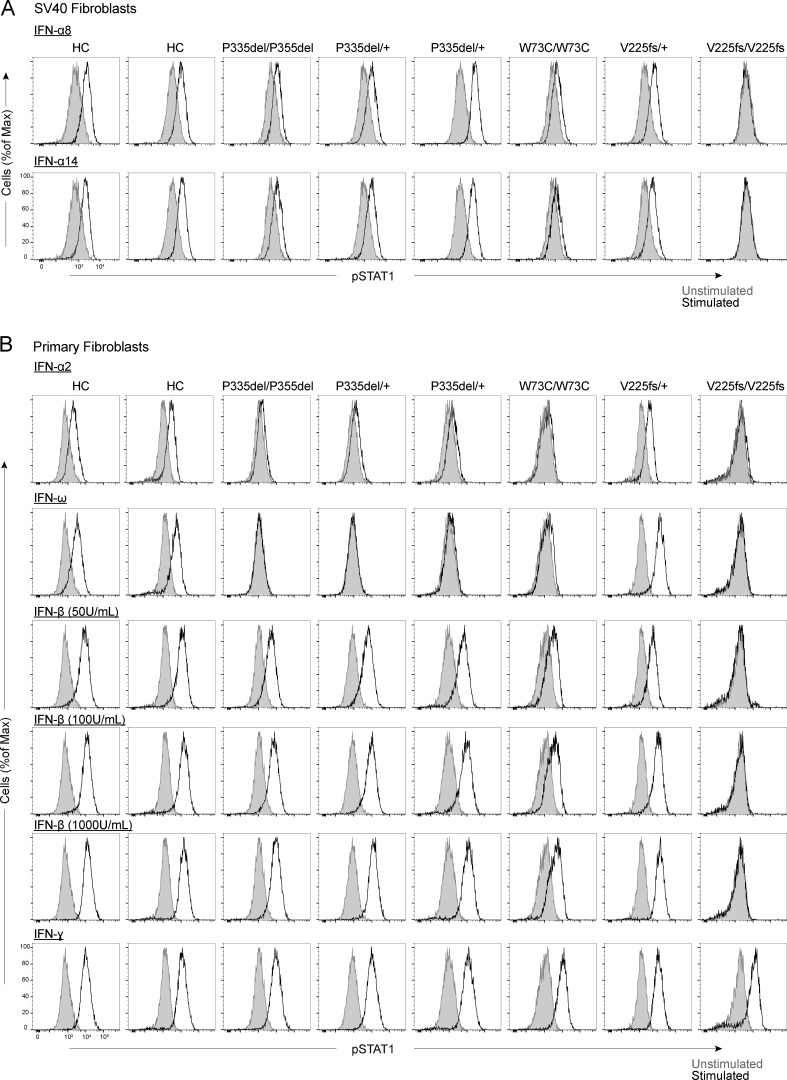
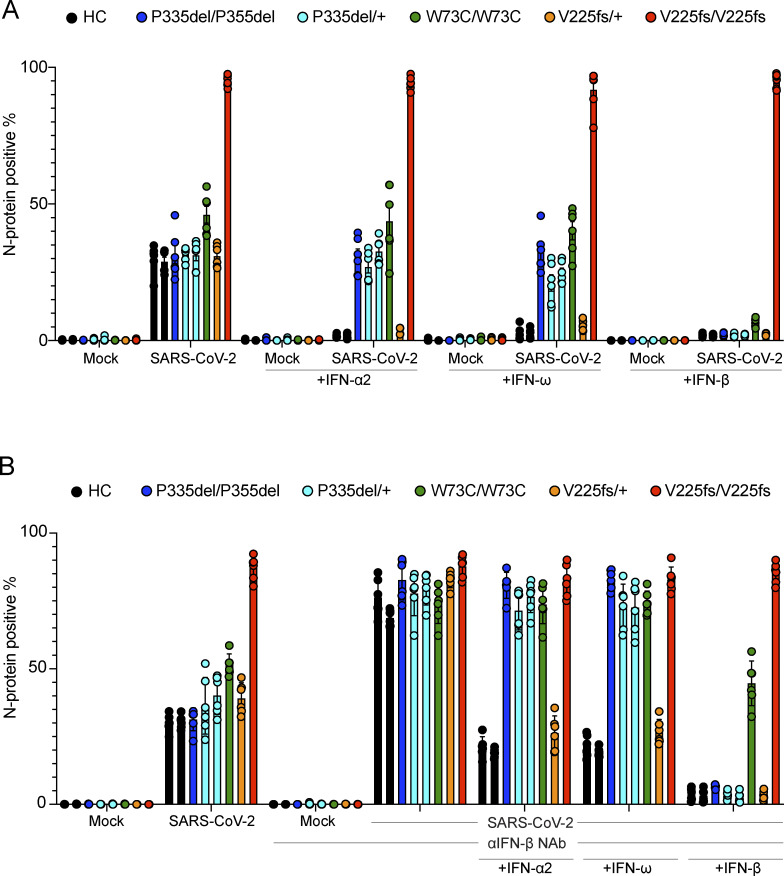
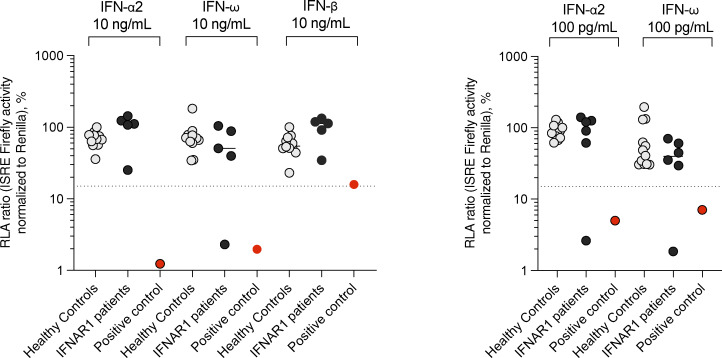
Autosomal recessive deficiency of the IFNAR1 or IFNAR2 chain of the human type I IFN receptor abolishes cellular responses to IFN-α, -β, and -ω, underlies severe viral diseases, and is globally very rare, except for IFNAR1 and IFNAR2 deficiency in Western Polynesia and the Arctic, respectively. We report 11 human IFNAR1 alleles, the products of which impair but do not abolish responses to IFN-α and -ω without affecting responses to IFN-β. Ten of these alleles are rare in all populations studied, but the remaining allele (P335del) is common in Southern China (minor allele frequency ≈2%). Cells heterozygous for these variants display a dominant phenotype in vitro with impaired responses to IFN-α and -ω, but not -β, and viral susceptibility. Negative dominance, rather than haploinsufficiency, accounts for this dominance. Patients heterozygous for these variants are prone to viral diseases, attesting to both the dominance of these variants clinically and the importance of IFN-α and -ω for protective immunity against some viruses.
© 2024 Al Qureshah et al.
Conflict of interest statement
Disclosures: N. de Prost reported personal fees from AstraZeneca and personal fees from Moderna outside the submitted work. J.-L. Casanova reported a patent to PCT/US2021/042741 pending. No other disclosures were reported.
Figures


References
-
- Abolhassani, H., Landegren N., Bastard P., Materna M., Modaresi M., Du L., Aranda-Guillén M., Sardh F., Zuo F., Zhang P., et al. 2022. Inherited IFNAR1 deficiency in a child with both critical COVID-19 pneumonia and multisystem inflammatory syndrome. J. Clin. Immunol. 42:471–483. 10.1007/s10875-022-01215-7 - DOI - PMC - PubMed
-
- Alotaibi, F., Alharbi N.K., Rosen L.B., Asiri A.Y., Assiri A.M., Balkhy H.H., Al Jeraisy M., Mandourah Y., AlJohani S., Al Harbi S., et al. 2023. Type I interferon autoantibodies in hospitalized patients with Middle East respiratory syndrome and association with outcomes and treatment effect of interferon beta-1b in MIRACLE clinical trial. Influenza Other Respir. Viruses. 17:e13116. 10.1111/irv.13116 - DOI - PMC - PubMed
-
- Armangué, T., Olivé-Cirera G., Martínez-Hernandez E., Rodes M., Peris-Sempere V., Guasp M., Ruiz R., Palou E., González A., Marcos M.Á., et al. 2023. Neurologic complications in herpes simplex encephalitis: Clinical, immunological and genetic studies. Brain. 146:4306–4319. 10.1093/brain/awad238 - DOI - PubMed
-
- Asano, T., Boisson B., Onodi F., Matuozzo D., Moncada-Velez M., Maglorius Renkilaraj M.R.L., Zhang P., Meertens L., Bolze A., Materna M., et al. 2021. X-linked recessive TLR7 deficiency in ∼1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6:eabl4348. 10.1126/sciimmunol.abl4348 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- EA20170638020/French Foundation for Medical Research
- King Abdulaziz City for Science and Technology
- Institut de Recherche pour le Développement
- Grandir - Fonds de solidarité pour l'enfance
- Battersea and Bowery Advisory Group
- Institut Pasteur
- King Saud University
- Imagine Institute
- R01 AI163029/AI/NIAID NIH HHS/United States
- Rockefeller University
- 12960/2021-2/Conselho Nacional de Desenvolvimento Científico e Tecnológico
- Meyer Foundation
- Fondation Bettencourt-Schueller
- COVID-1026207/American Lung Association
- UL1 TR001866/TR/NCATS NIH HHS/United States
- UL1TR001866/NH/NIH HHS/United States
- Institut National de la Santé et de la Recherche Médicale
- French Ministry of Higher Education, Research, and Innovation
- HHMI/Howard Hughes Medical Institute/United States
- Square Foundation
- Fondation du Souffle
- SCOR Corporate Foundation for Science
- King Abdullah University of Science and Technology
- St. Giles Foundation
- WT_/Wellcome Trust/United Kingdom
- Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil
- General Atlantic Foundation
- ANR-10-IAHU-01/French National Research Agency
- Paris Cité University
- 824110/European Union's Horizon 2020 research and innovation program
- 101057100/HORIZON-HLTH-2021-DISEASE-04 program
- JPB Foundation
- RR/NCRR NIH HHS/United States
- Stavros Niarchos Foundation
- Fisher Center for Alzheimer's Research Foundation
LinkOut - more resources
Full Text Sources
