

Engineering Phages to Fight Multidrug-Resistant Bacteria
- PMID: 39680919
- PMCID: PMC11758799
- DOI: 10.1021/acs.chemrev.4c00681
Engineering Phages to Fight Multidrug-Resistant Bacteria
Abstract
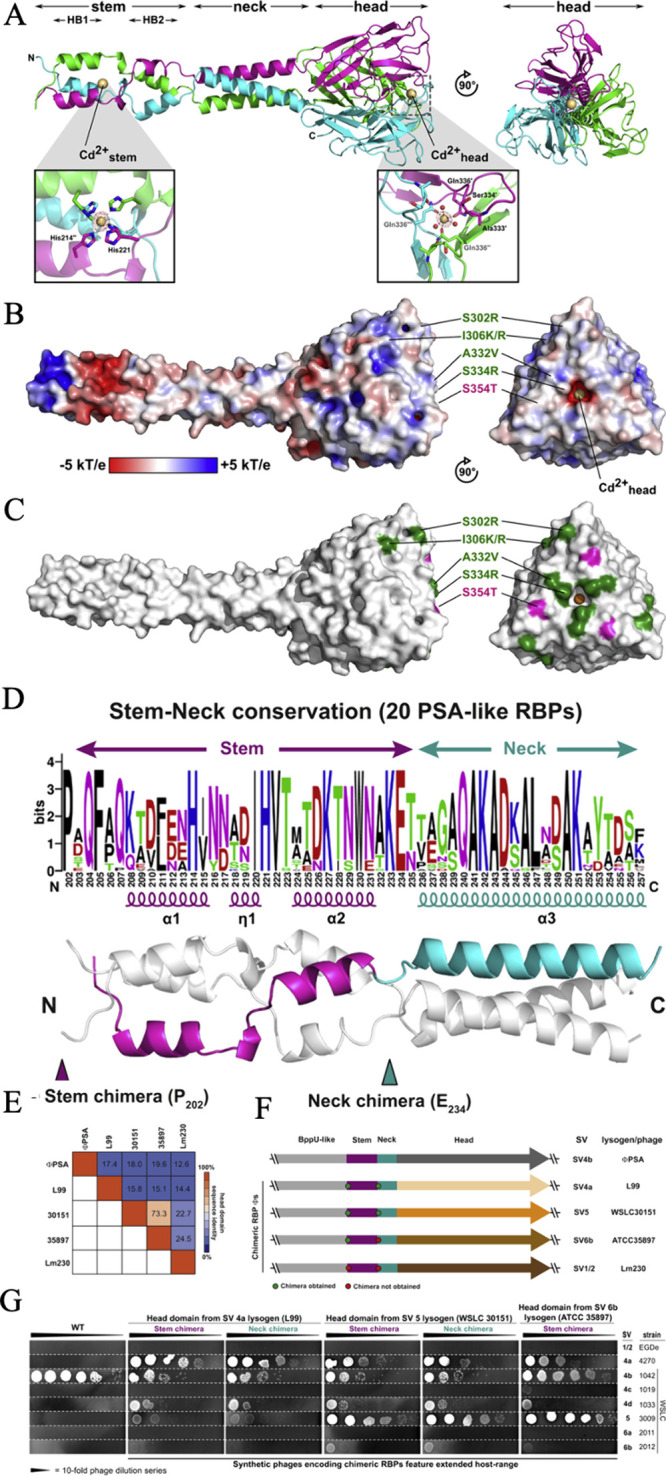
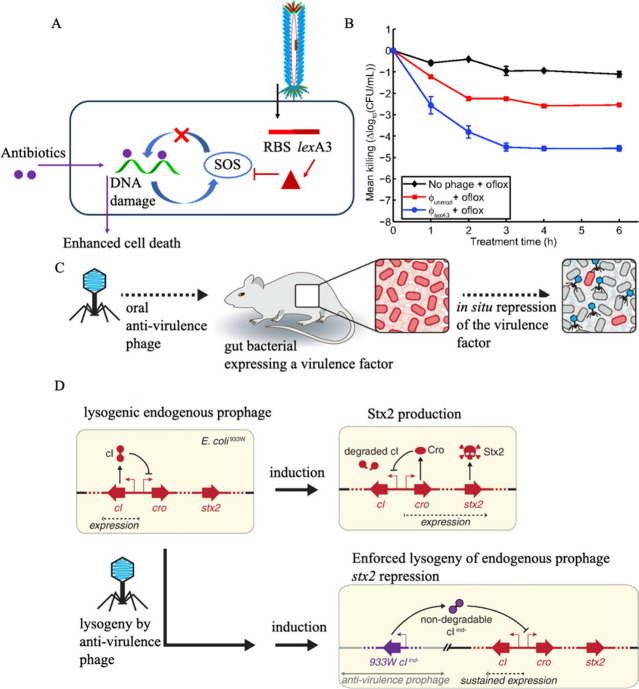
Facing the global "superbug" crisis due to the emergence and selection for antibiotic resistance, phages are among the most promising solutions. Fighting multidrug-resistant bacteria requires precise diagnosis of bacterial pathogens and specific cell-killing. Phages have several potential advantages over conventional antibacterial agents such as host specificity, self-amplification, easy production, low toxicity as well as biofilm degradation. However, the narrow host range, uncharacterized properties, as well as potential risks from exponential replication and evolution of natural phages, currently limit their applications. Engineering phages can not only enhance the host bacteria range and improve phage efficacy, but also confer new functions. This review first summarizes major phage engineering techniques including both chemical modification and genetic engineering. Subsequent sections discuss the applications of engineered phages for bacterial pathogen detection and ablation through interdisciplinary approaches of synthetic biology and nanotechnology. We discuss future directions and persistent challenges in the ongoing exploration of phage engineering for pathogen control.
Conflict of interest statement
The authors declare no competing financial interest.
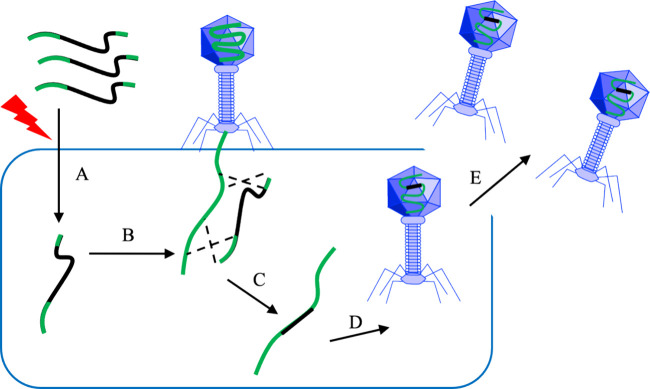
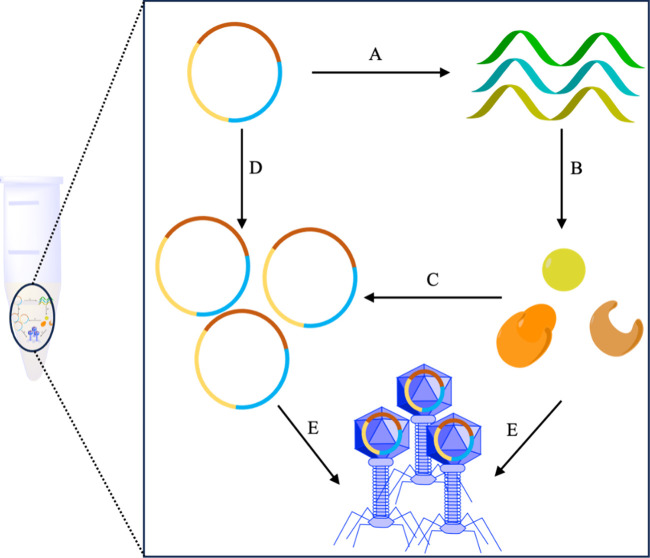
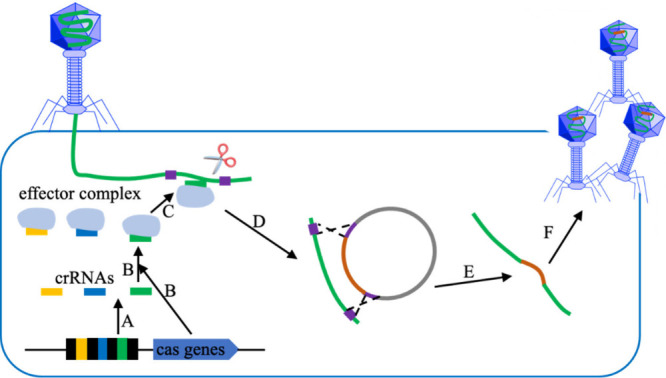
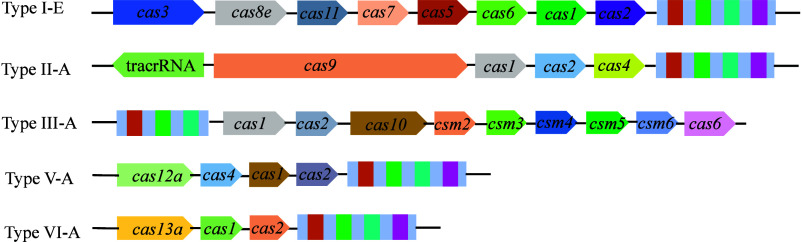
Figures












References
-
- Ikuta K. S.; Swetschinski L. R.; Robles Aguilar G.; Sharara F.; Mestrovic T.; Gray A. P.; Davis Weaver N.; Wool E. E.; Han C.; Gershberg Hayoon A.; et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. 10.1016/S0140-6736(22)02185-7. - DOI - PMC - PubMed
-
- Beyer P.; Paulin S. The Antibacterial Research and Development Pipeline Needs Urgent Solutions. ACS Infect. Dis. 2020, 6, 1289–1291. 10.1021/acsinfecdis.0c00044. - DOI
