

The p.R66W Variant in RAC3 Causes Severe Fetopathy Through Variant-Specific Mechanisms
- PMID: 39682779
- PMCID: PMC11640247
- DOI: 10.3390/cells13232032
The p.R66W Variant in RAC3 Causes Severe Fetopathy Through Variant-Specific Mechanisms
Abstract
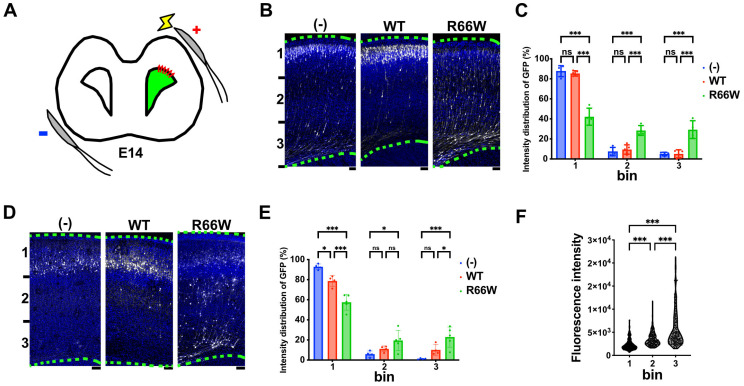
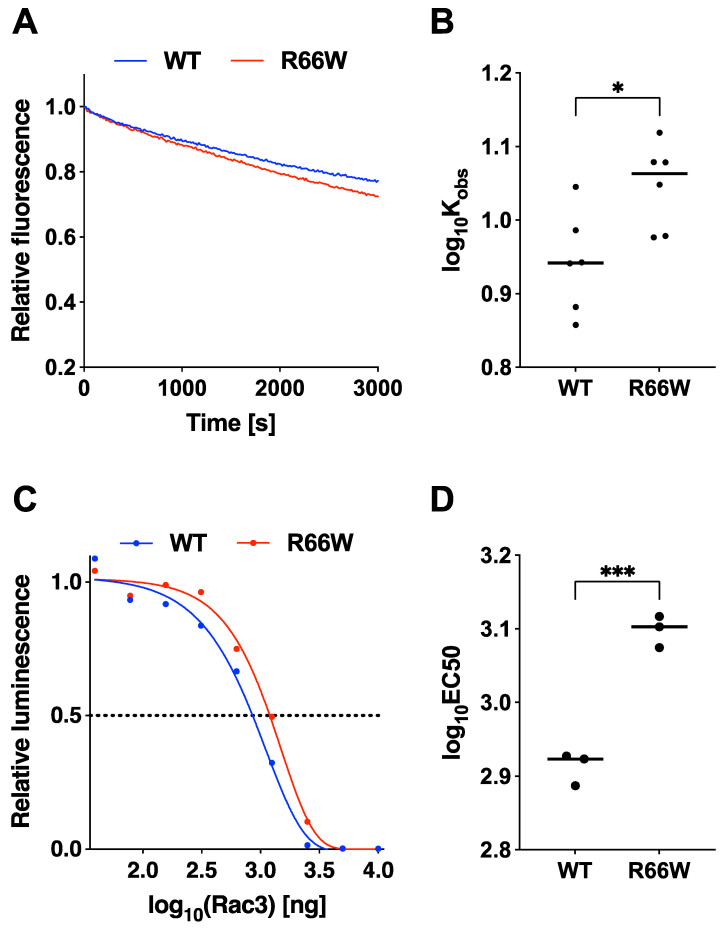
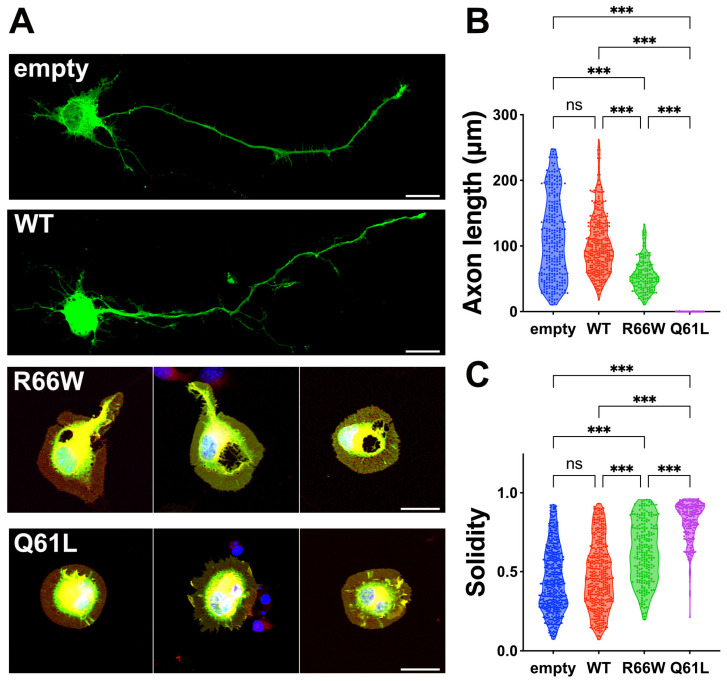
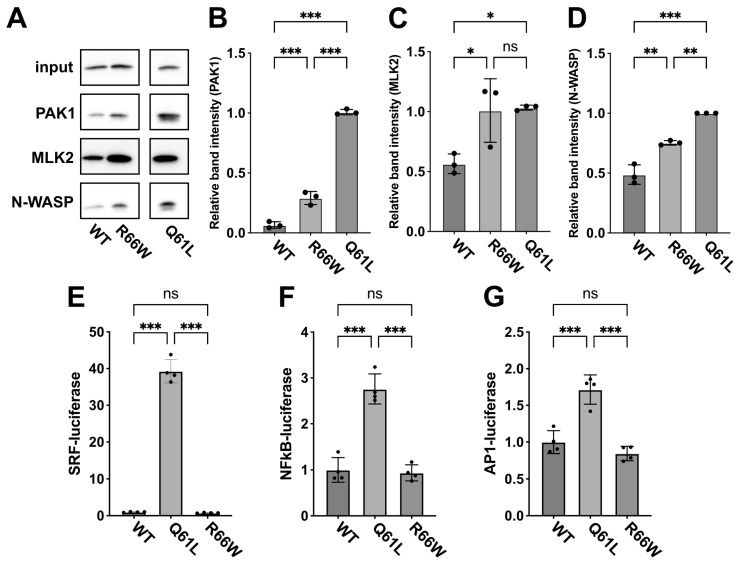
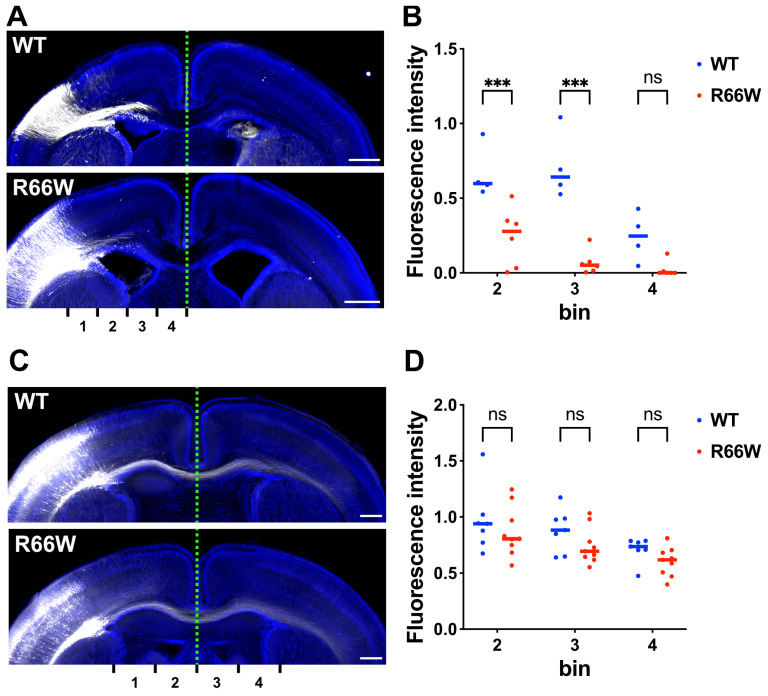
RAC3 encodes a small GTPase of the Rho family that plays a critical role in actin cytoskeleton remodeling and intracellular signaling regulation. Pathogenic variants in RAC3, all of which reported thus far affect conserved residues within its functional domains, have been linked to neurodevelopmental disorders characterized by diverse phenotypic features, including structural brain anomalies and facial dysmorphism (NEDBAF). Recently, a novel de novo RAC3 variant (NM_005052.3): c.196C>T, p.R66W was identified in a prenatal case with fetal akinesia deformation sequence (a spectrum of conditions that interfere with the fetus's ability to move), and complex brain malformations featuring corpus callosum agenesis, diencephalosynapsis, kinked brainstem, and vermian hypoplasia. To investigate the mechanisms underlying the association between RAC3 deficiency and this unique, distinct clinical phenotype, we explored the pathophysiological significance of the p.R66W variant in brain development. Biochemical assays revealed a modest enhancement in intrinsic GDP/GTP exchange activity and an inhibitory effect on GTP hydrolysis. Transient expression studies in COS7 cells demonstrated that RAC3-R66W interacts with the downstream effectors PAK1, MLK2, and N-WASP but fails to activate SRF-, AP1-, and NFkB-mediated transcription. Additionally, overexpression of RAC3-R66W significantly impaired differentiation in primary cultured hippocampal neurons. Acute expression of RAC3-R66W in vivo by in utero electroporation resulted in impairments in cortical neuron migration and axonal elongation during corticogenesis. Collectively, these findings suggest that the p.R66W variant may function as an activated version in specific signaling pathways, leading to a distinctive and severe prenatal phenotype through variant-specific mechanisms.
Keywords: RAC3; axon extension; corticogenesis; neurodevelopmental disorder; neuronal migration; small GTPase.
Conflict of interest statement
All authors declare no conflicts of interest.
Figures
Similar articles
-
A p.N92K variant of the GTPase RAC3 disrupts cortical neuron migration and axon elongation.J Biol Chem. 2025 Apr;301(4):108346. doi: 10.1016/j.jbc.2025.108346. Epub 2025 Feb 25. J Biol Chem. 2025. PMID: 40015633 Free PMC article.
-
Gain-of-function p.F28S variant in RAC3 disrupts neuronal differentiation, migration and axonogenesis during cortical development, leading to neurodevelopmental disorder.J Med Genet. 2023 Mar;60(3):223-232. doi: 10.1136/jmedgenet-2022-108483. Epub 2022 May 20. J Med Genet. 2023. PMID: 35595279
-
Variant-specific changes in RAC3 function disrupt corticogenesis in neurodevelopmental phenotypes.Brain. 2022 Sep 14;145(9):3308-3327. doi: 10.1093/brain/awac106. Brain. 2022. PMID: 35851598 Free PMC article.
-
The Rac3 GTPase in Neuronal Development, Neurodevelopmental Disorders, and Cancer.Cells. 2019 Sep 11;8(9):1063. doi: 10.3390/cells8091063. Cells. 2019. PMID: 31514269 Free PMC article. Review.
-
Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What's behind Neuro-RACopathies.Cells. 2021 Dec 2;10(12):3395. doi: 10.3390/cells10123395. Cells. 2021. PMID: 34943902 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
