

The Neuroprotective Effect of the X Protein of Orthobornavirus Bornaense Type 1 in Amyotrophic Lateral Sclerosis
- PMID: 39684507
- PMCID: PMC11641280
- DOI: 10.3390/ijms252312789
The Neuroprotective Effect of the X Protein of Orthobornavirus Bornaense Type 1 in Amyotrophic Lateral Sclerosis
Abstract
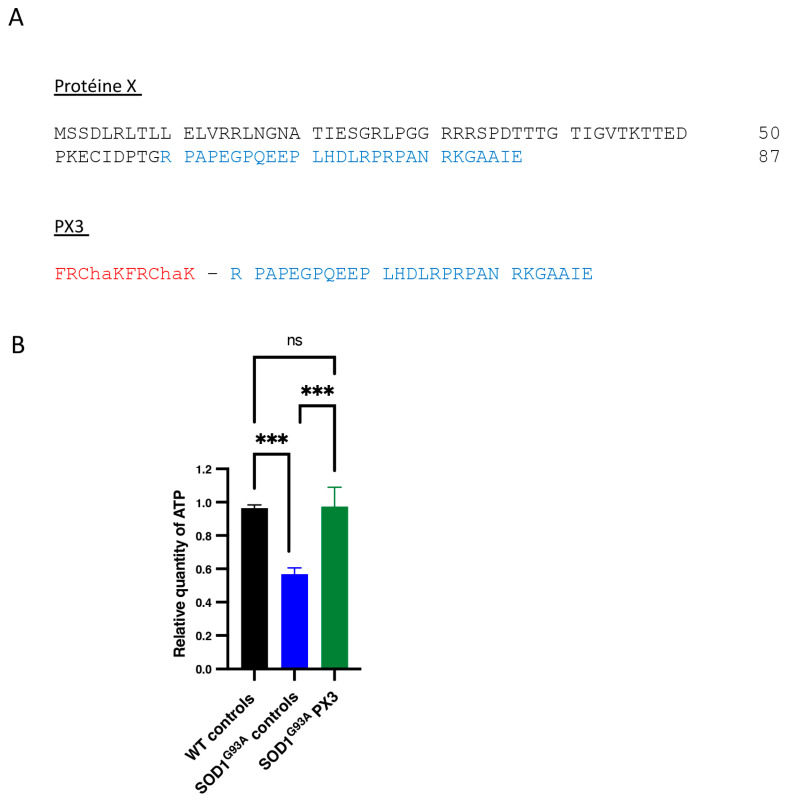
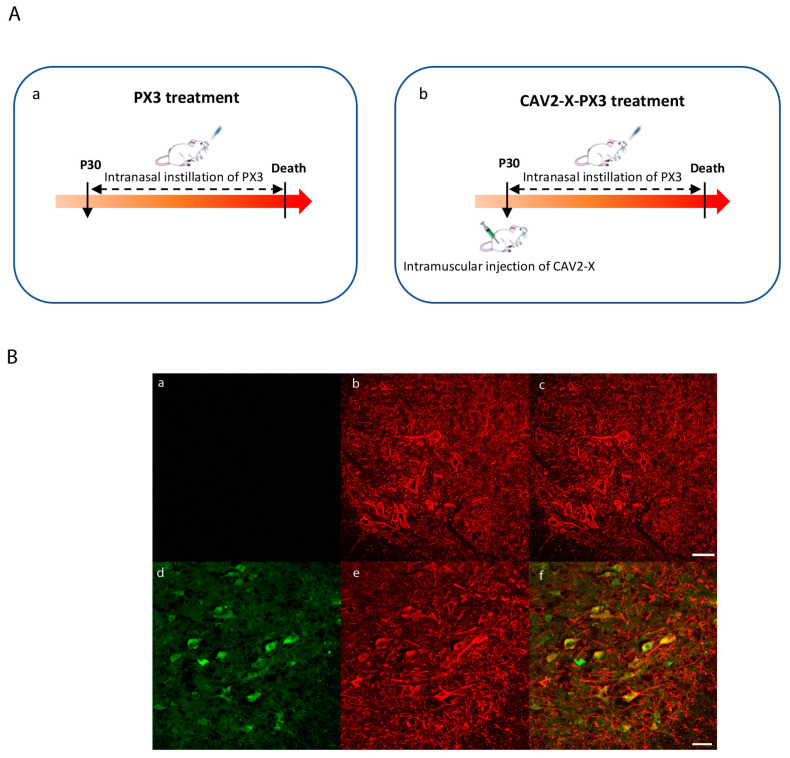
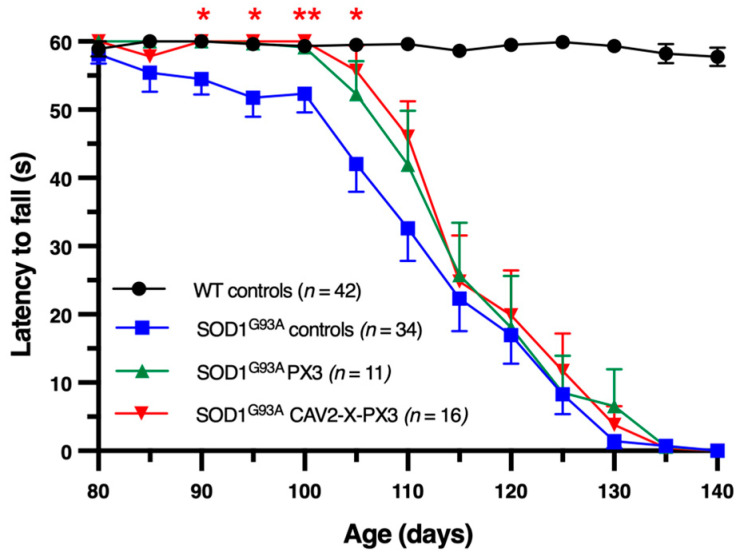
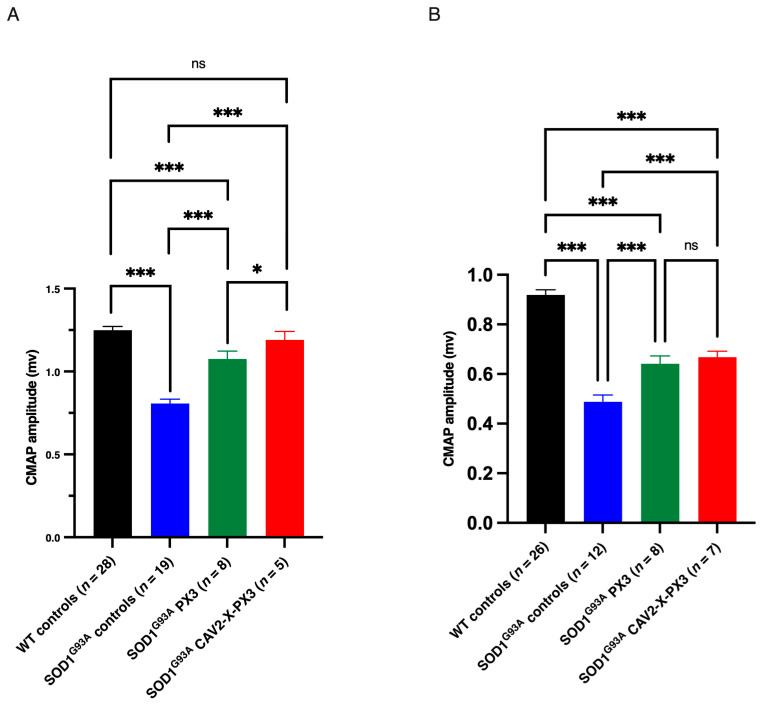
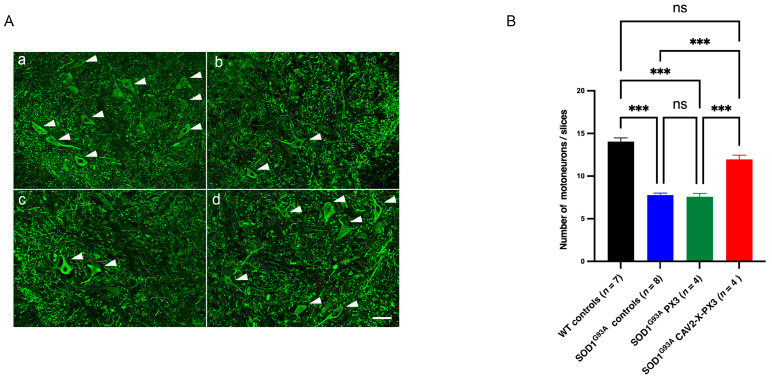
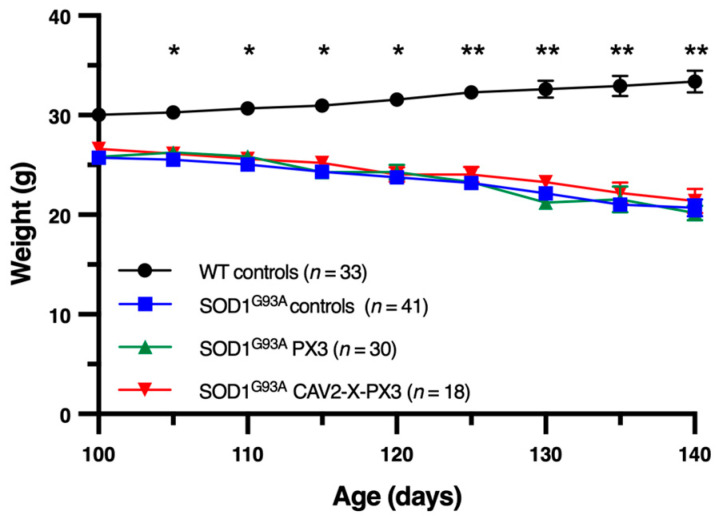
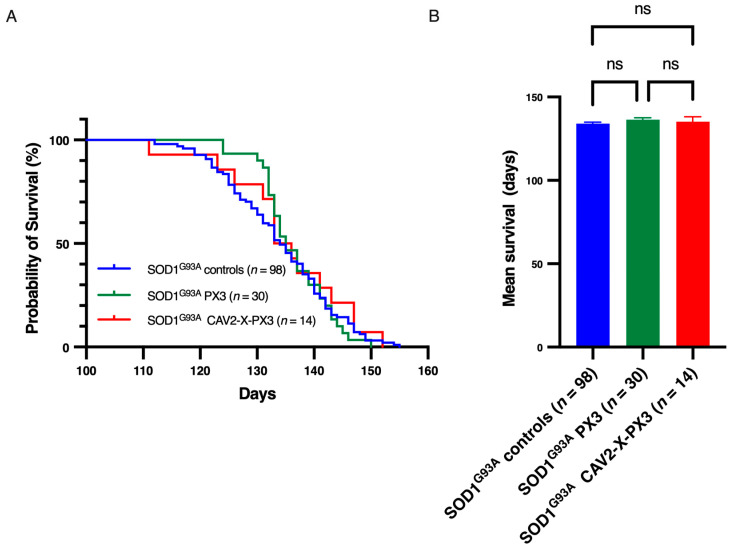
In amyotrophic lateral sclerosis (ALS), early mitochondrial dysfunction may contribute to progressive motor neuron loss. Remarkably, the ectopic expression of the Orthobornavirus bornaense type 1 (BoDV-1) X protein in mitochondria blocks apoptosis and protects neurons from degeneration. Therefore, this study examines the neuroprotective effects of X protein in an ALS mouse model. We first tested in vitro the effect of the X-derived peptide (PX3) on motoneurons primary cultures of SOD1G93A mice. The total intracellular adenosine triphosphate (ATP) content was measured after incubation of the peptide. We next tested in vivo the intramuscular injection of X protein using a canine viral vector (CAV2-X) and PX3 intranasal administrations in SOD1G93A mice. Disease onset and progression were assessed through rotarod performance, functional motor unit analysis via electrophysiology, and motor neuron survival by immunohistochemistry. The results showed that in vitro PX3 restored the ATP level in SOD1G93A motor neurons. In vivo, treated mice demonstrated better motor performance, preserved motor units, and higher motor neuron survival. Although life expectancy was not extended in this severe mouse model of motor neuron degeneration, the present findings clearly demonstrate the neuroprotective potential of X protein in a model of ALS. We are convinced that further studies may improve the therapeutic impact of X protein with optimized administration methods.
Keywords: ALS; BoDV-1; SOD1G93A mice; X protein; mitochondria.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
MeSH terms
Substances
Grants and funding
- Evaluation of the therapeutic potential of the Borna virus X protein and X-derived peptides in ALS/Association pour la recherche sur la Sclérose Latérale Amyotrophique
- Award Fabrice Le Mouaher 2020/Fondation pour la Recherche Médicale
- trampoline grant 20219/Association Francaise contre les Myopathies
- X protein and ALS/Rotary Club of Bergerac
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
