

AXL-TBK1 driven AKT3 activation promotes metastasis
- PMID: 39689180
- PMCID: PMC12229235
- DOI: 10.1126/scisignal.ado6057
AXL-TBK1 driven AKT3 activation promotes metastasis
Abstract
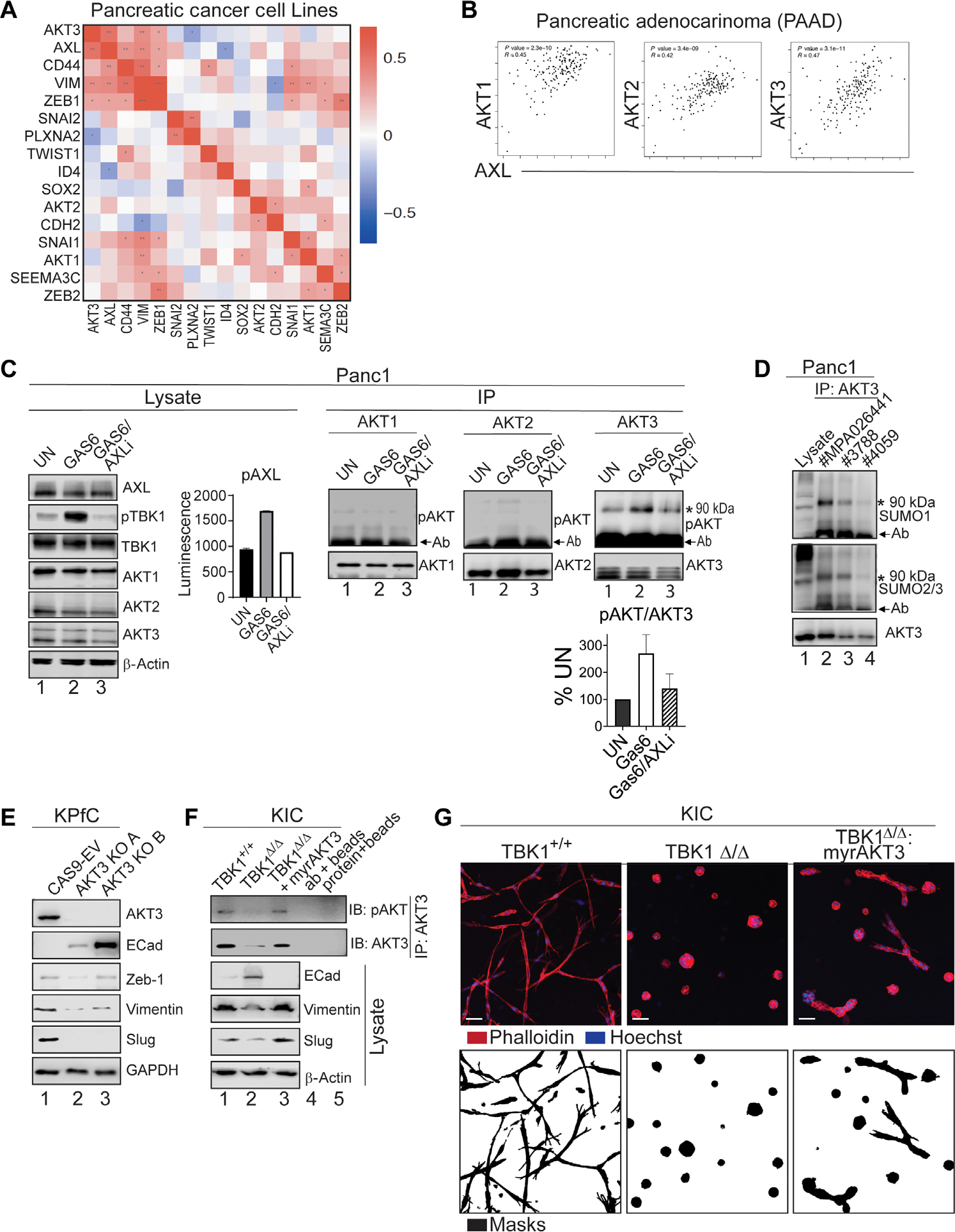
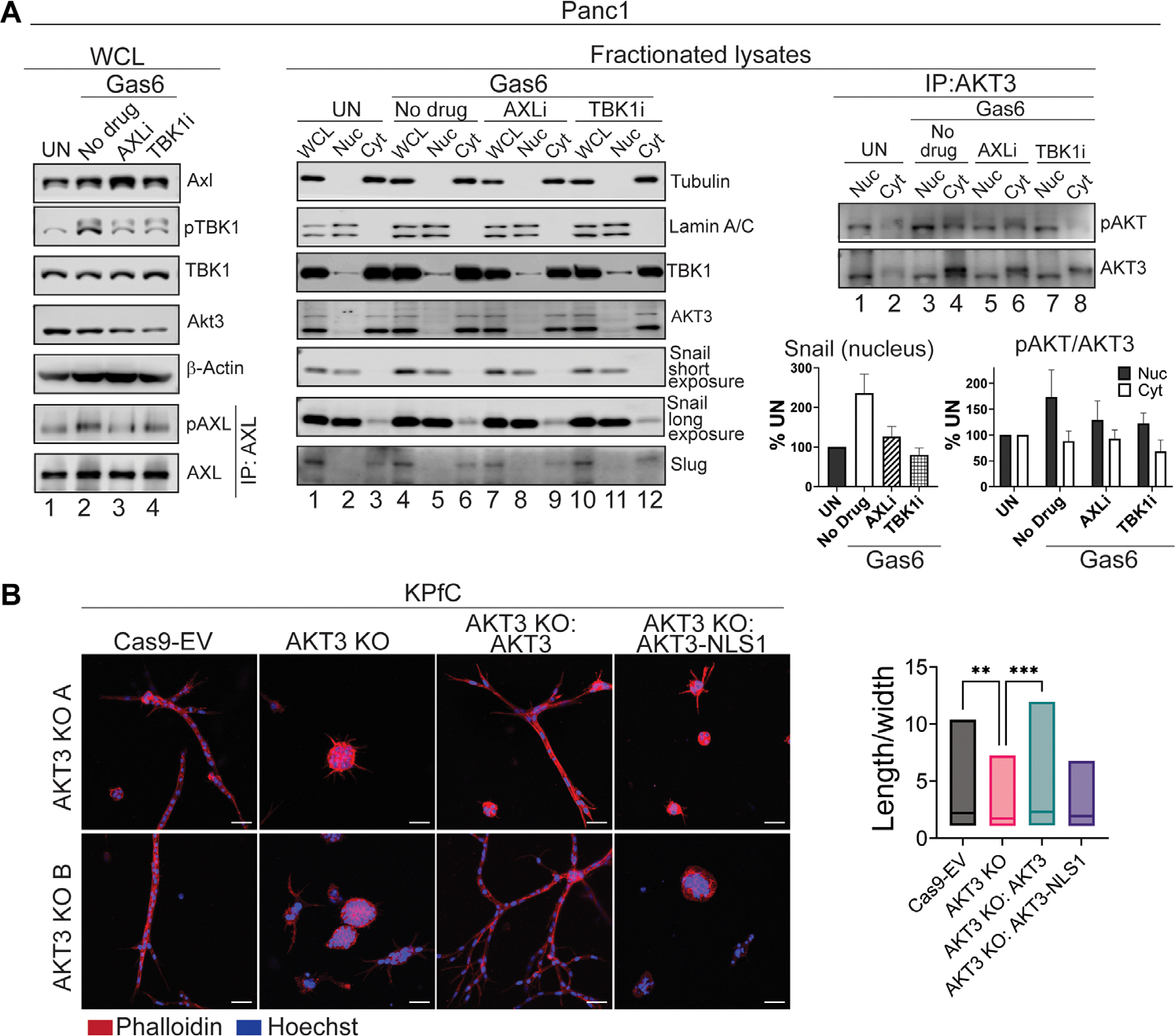
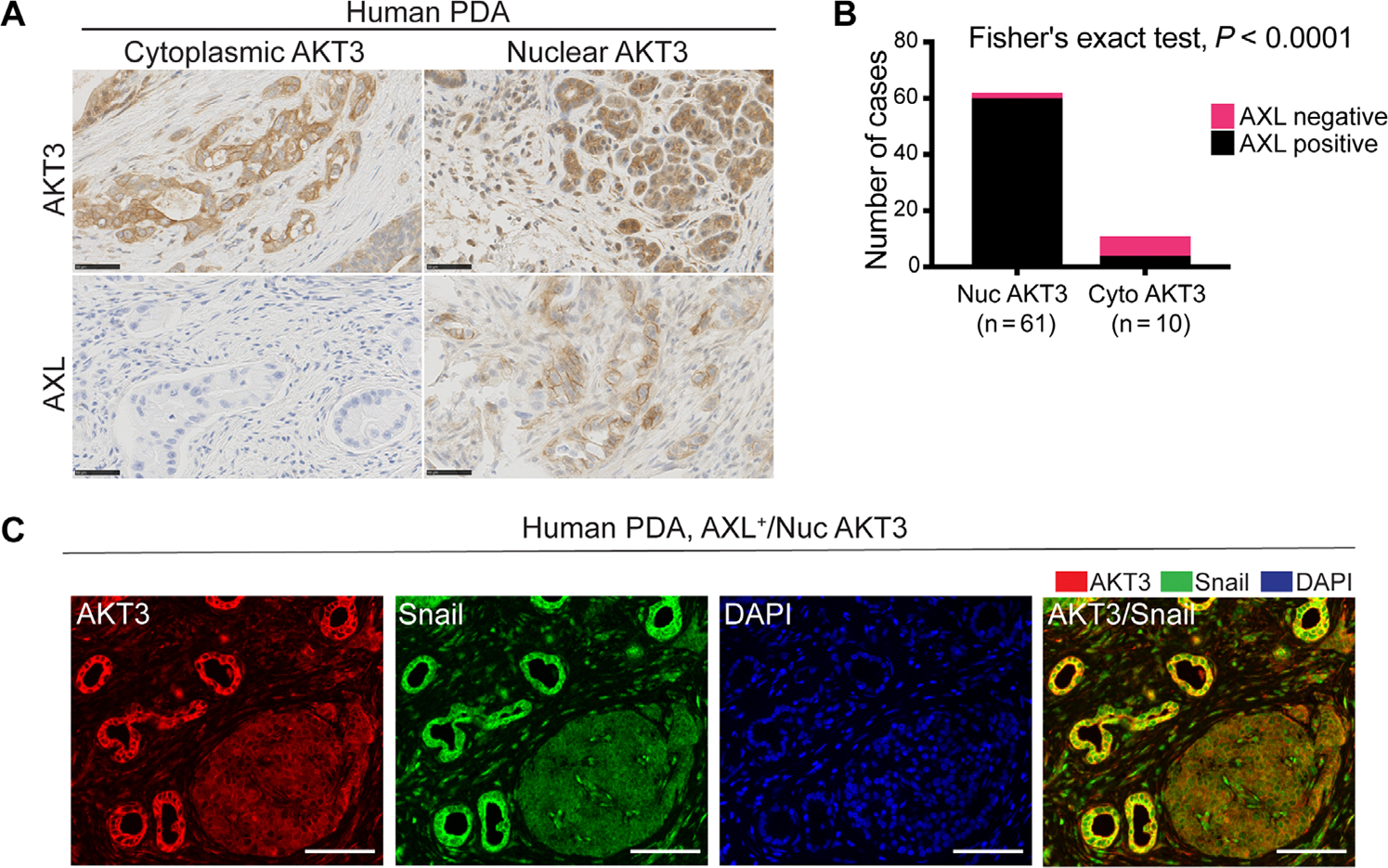
The receptor tyrosine kinase AXL promotes tumor progression, metastasis, and therapy resistance through the induction of epithelial-mesenchymal transition (EMT). Here, we found that activation of AXL resulted in the phosphorylation of TANK-binding kinase 1 (TBK1) and the downstream activation of AKT3 and Snail, a transcription factor critical for EMT. Mechanistically, we showed that TBK1 directly bound to and phosphorylated AKT3 in a manner dependent on the multiprotein complex mTORC1. Upon activation, AKT3 interacted with and promoted the nuclear accumulation of Snail, which led to increased EMT as assessed by marker abundance. In human pancreatic ductal adenocarcinoma tissue, nuclear AKT3 colocalized with Snail and correlated with worse clinical outcomes. Primary mouse pancreatic cancer cells deficient in AKT3 showed reduced metastatic spread in vivo, suggesting selective AKT3 inhibition as a potential therapeutic avenue for targeting EMT in aggressive cancers.
Conflict of interest statement
Figures





References
-
- Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH, Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 8, 27–33 (2011). - PubMed
-
- Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y, Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 15, 195–206 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
