

Indirect Formation of Peptide Bonds as a Prelude to Ribosomal Transpeptidation
- PMID: 39693575
- PMCID: PMC11726440
- DOI: 10.1021/jacs.4c10326
Indirect Formation of Peptide Bonds as a Prelude to Ribosomal Transpeptidation
Abstract
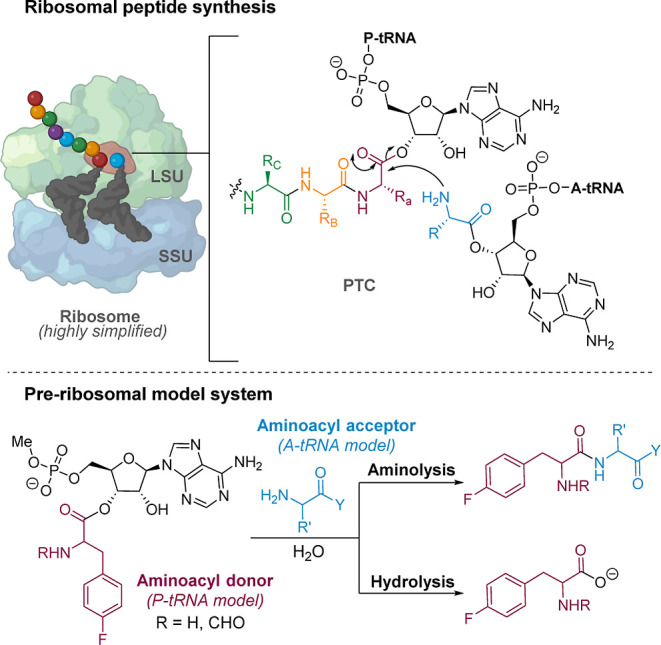
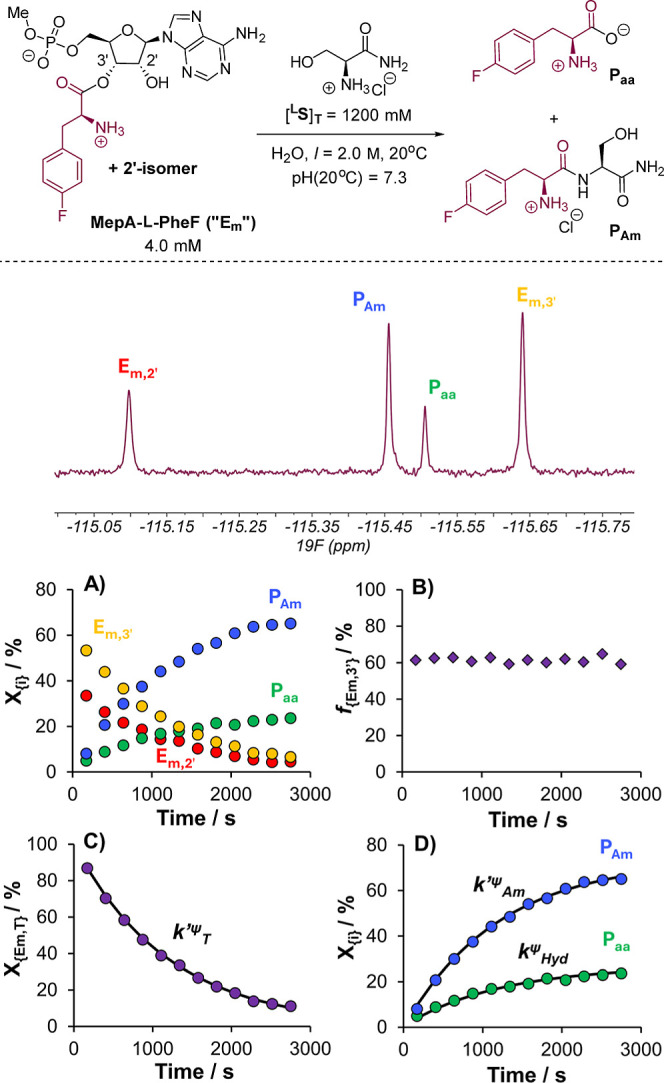
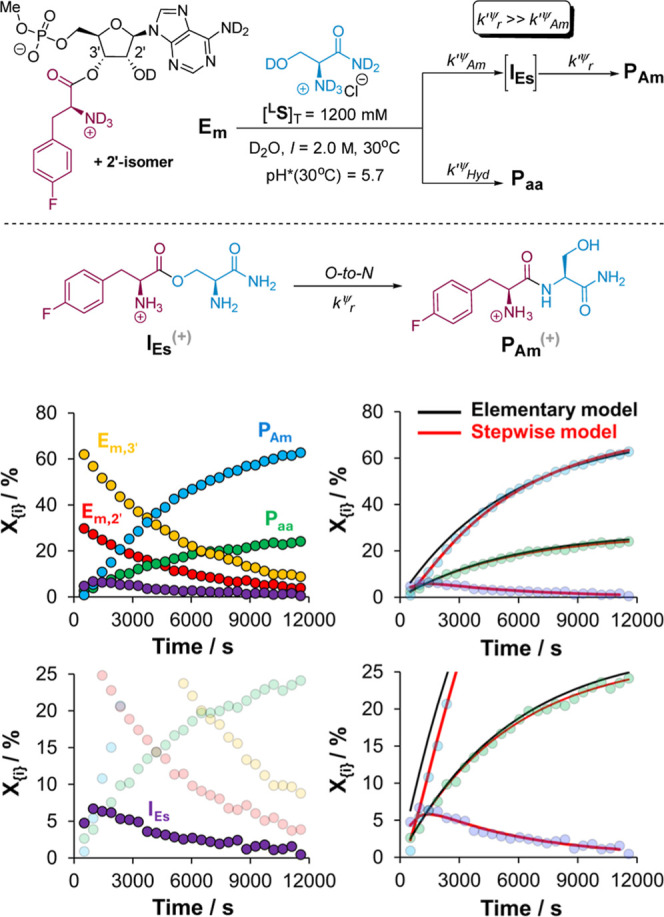
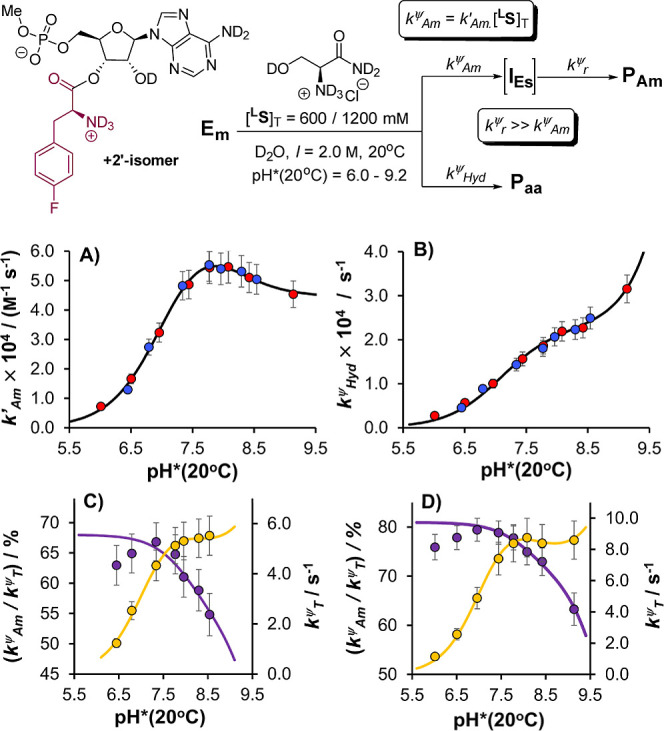
The catalytic competency of the ribosome in extant protein biosynthesis is thought to arise primarily from two sources: an ability to precisely juxtapose the termini of two key substrates─3'-aminoacyl and N-acyl-aminoacyl tRNAs─and an ability to ease direct transpeptidation by their desolvation and encapsulation. In the absence of ribosomal, or enzymatic, protection, however, these activated alkyl esters undergo efficient hydrolysis, while significant entropic barriers serve to hamper their intermolecular cross-aminolysis in bulk water. Given that the spontaneous emergence of a catalyst of comparable size and sophistication to the ribosome in a prebiotic RNA world would appear implausible, it is thus natural to ask how appreciable peptide formation could have occurred with such substrates in bulk water without the aid of advanced ribozymatic catalysis. Using a combination of fluorine-tagged aminoacyl adenylate esters, in situ monitoring by 19F{1H} NMR spectroscopy, analytical deconvolution of kinetics, pH-rate profile analysis, and temperature-dependence studies, we here explore the mechanistic landscape of indirect amidation, via transesterification and O-to-N rearrangement, as a highly efficient, alternative manifold for transpeptidation that may have served as a prelude to ribosomal peptide synthesis. Our results suggest a potentially overlooked role for those amino acids implicated by the cyanosulfidic reaction network with hydroxyl side chains (Ser and Thr), and they also help to resolve some outstanding ambiguities in the broader literature regarding studies of similar systems (e.g., aminolyzes with Tris buffer). The evolutionary implications of this mode of peptide synthesis and the involvement of a very specific subset of amino acids are discussed.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
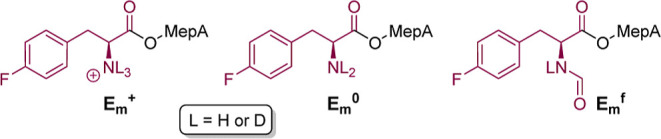
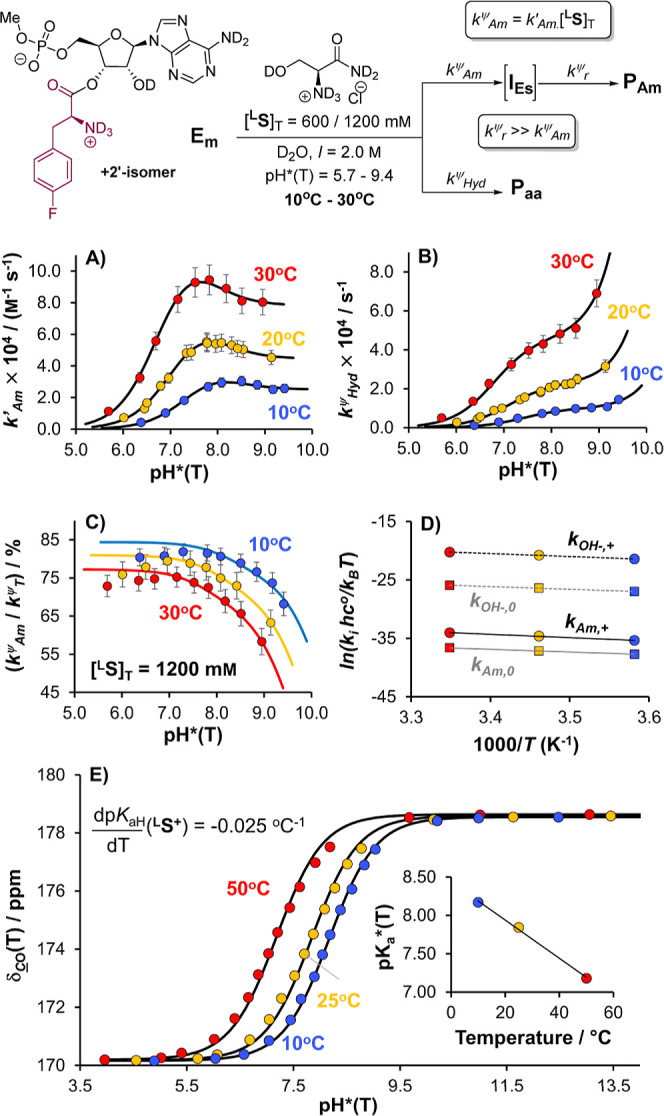
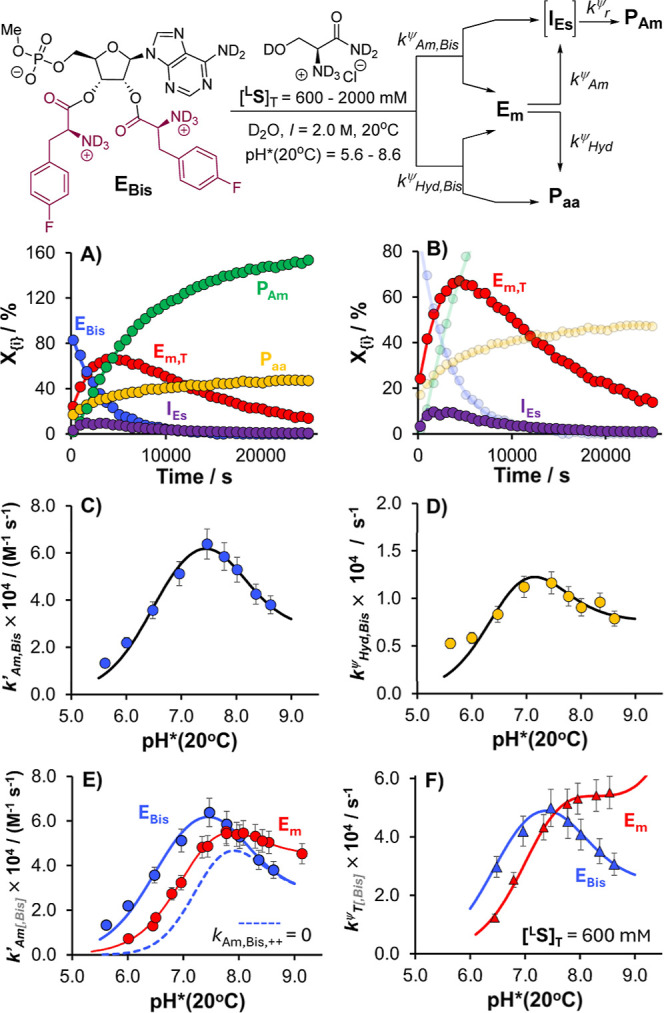
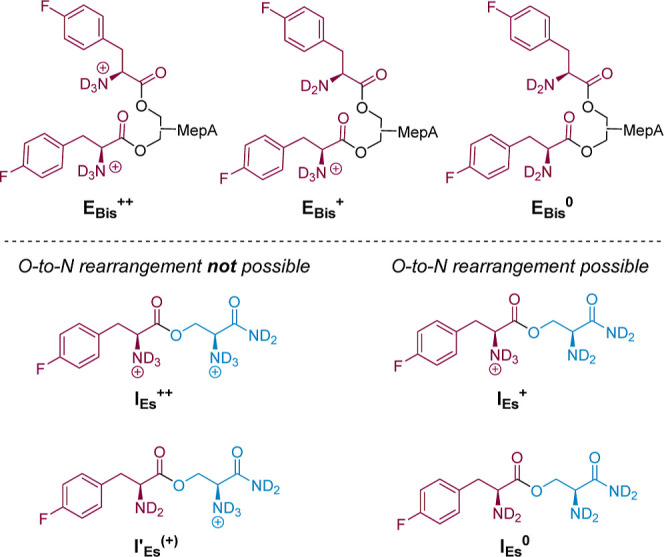
References
-
- Ban N.; Nissen P.; Hansen J.; Moore P. B.; Steitz T. A. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Å Resolution. Science 2000, 289, 905–920. 10.1126/science.289.5481.905. - DOI - PubMed
- Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. 10.1126/science.289.5481.920. - DOI - PubMed
- Schlünzen F.; Zarivach R.; Harms J.; Bashan A.; Tocilj A.; Albrecht R.; Yonath A.; Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. 10.1038/35101544. - DOI - PubMed
- Harms J.; Schluenzen F.; Zarivach R.; Bashan A.; Gat S.; Agmon I.; Bartels H.; Franceschi F.; Yonath A. High Resolution Structure of the Large Ribosomal Subunit from a Mesophilic Eubacterium. Cell 2001, 107, 679–688. 10.1016/S0092-8674(01)00546-3. - DOI - PubMed
- Hansen J. L.; Schmeing T. M.; Moore P. B.; Steitz T. A. Structural insights into peptide bond formation. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 11670–11675. 10.1073/pnas.172404099. - DOI - PMC - PubMed
- Beringer M.; Bruell C.; Xiong L.; Pfister P.; Bieling P.; Katunin V. I.; Mankin A. S.; Böttger E. C.; Rodnina M. V. Essential Mechanisms in the Catalysis of Peptide Bond Formation on the Ribosome. J. Biol. Chem. 2005, 280, 36065–36072. 10.1074/jbc.M507961200. - DOI - PubMed
- Trobro S.; Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12395–12400. 10.1073/pnas.0504043102. - DOI - PMC - PubMed
- Bowman J. C.; Petrov A. S.; Frenkel-Pinter M.; Penev P. I.; Williams L. D. Root of the Tree: The Significance, Evolution, and Origins of the Ribosome. Chem. Rev. 2020, 120, 4848–4878. 10.1021/acs.chemrev.9b00742. - DOI - PubMed
-
- Polacek N.; Mankin A. S. The Ribosomal Peptidyl Transferase Center: Structure, Function, Evolution, Inhibition. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 285–311. 10.1080/10409230500326334. - DOI - PubMed
- Beringer M.; Rodnina M. V. The Ribosomal Peptidyl Transferase. Mol. Cell 2007, 26, 311–321. 10.1016/j.molcel.2007.03.015. - DOI - PubMed
-
- Noller H. F. Ribosomal RNA and Translation. Annu. Rev. Biochem. 1991, 60, 191–227. 10.1146/annurev.bi.60.070191.001203. - DOI - PubMed
- Green R.; Noller H. F. Ribosomes and Translation. Annu. Rev. Biochem. 1997, 66, 679–716. 10.1146/annurev.biochem.66.1.679. - DOI - PubMed
- Davidovich C.; Belousoff M.; Wekselman I.; Shapira T.; Krupkin M.; Zimmerman E.; Bashan A.; Yonath A. The Proto-Ribosome: An Ancient Nano-machine for Peptide Bond Formation. Isr. J. Chem. 2010, 50, 29–35. 10.1002/ijch.201000012. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
