

Nucleosome fibre topology guides transcription factor binding to enhancers
- PMID: 39695228
- PMCID: PMC11798873
- DOI: 10.1038/s41586-024-08333-9
Nucleosome fibre topology guides transcription factor binding to enhancers
Abstract
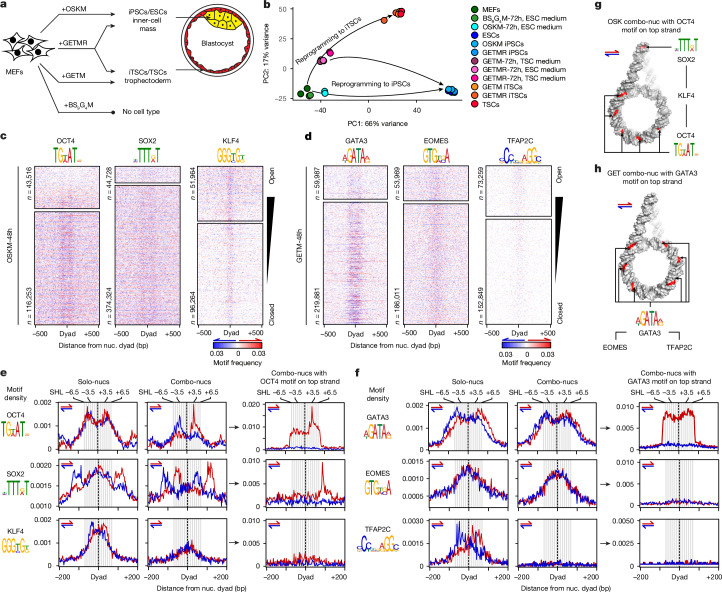
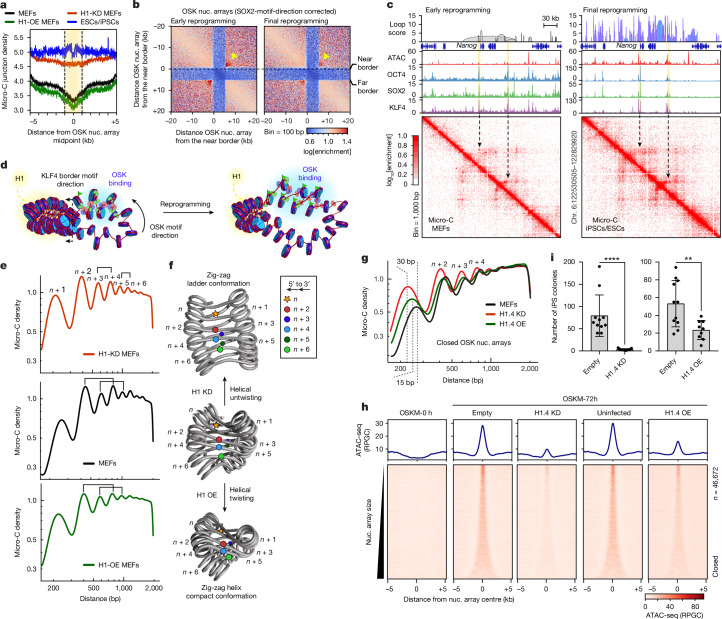
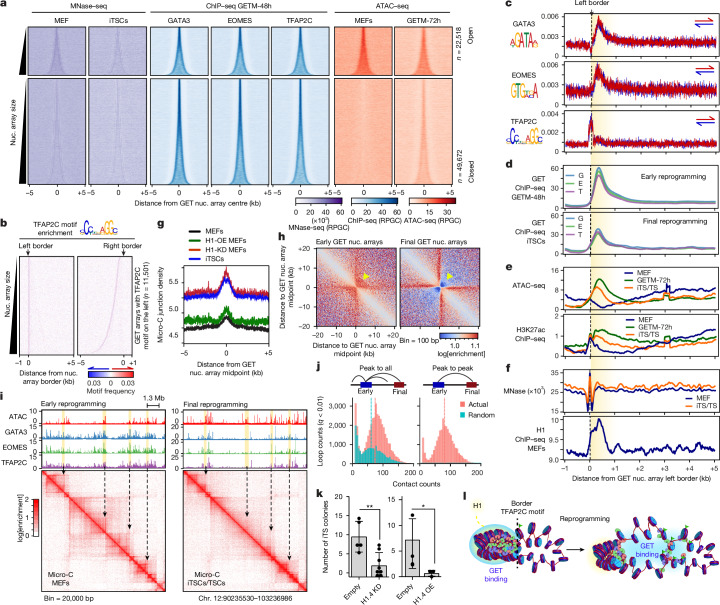
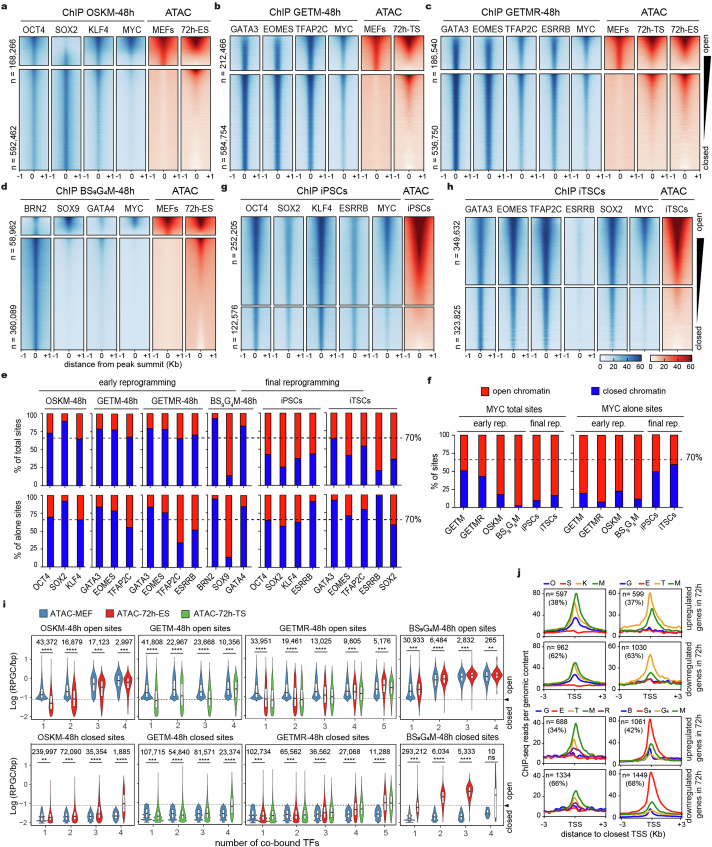
Cellular identity requires the concerted action of multiple transcription factors (TFs) bound together to enhancers of cell-type-specific genes. Despite TFs recognizing specific DNA motifs within accessible chromatin, this information is insufficient to explain how TFs select enhancers1. Here we compared four different TF combinations that induce different cell states, analysing TF genome occupancy, chromatin accessibility, nucleosome positioning and 3D genome organization at the nucleosome resolution. We show that motif recognition on mononucleosomes can decipher only the individual binding of TFs. When bound together, TFs act cooperatively or competitively to target nucleosome arrays with defined 3D organization, displaying motifs in particular patterns. In one combination, motif directionality funnels TF combinatorial binding along chromatin loops, before infiltrating laterally to adjacent enhancers. In other combinations, TFs assemble on motif-dense and highly interconnected loop junctions, and subsequently translocate to nearby lineage-specific sites. We propose a guided-search model in which motif grammar on nucleosome fibres acts as signpost elements, directing TF combinatorial binding to enhancers.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures











References
-
- Spitz, F. & Furlong, E. E. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet.13, 613–626 (2012). - PubMed
-
- Xu, J., Du, Y. & Deng, H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell16, 119–134 (2015). - PubMed
-
- Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell126, 663–676 (2006). - PubMed
-
- Benchetrit, H. et al. Extensive nuclear reprogramming underlies lineage conversion into functional trophoblast stem-like cells. Cell Stem Cell17, 543–556 (2015). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
