

Lithocholic acid binds TULP3 to activate sirtuins and AMPK to slow down ageing
- PMID: 39695235
- PMCID: PMC12222023
- DOI: 10.1038/s41586-024-08348-2
Lithocholic acid binds TULP3 to activate sirtuins and AMPK to slow down ageing
Abstract
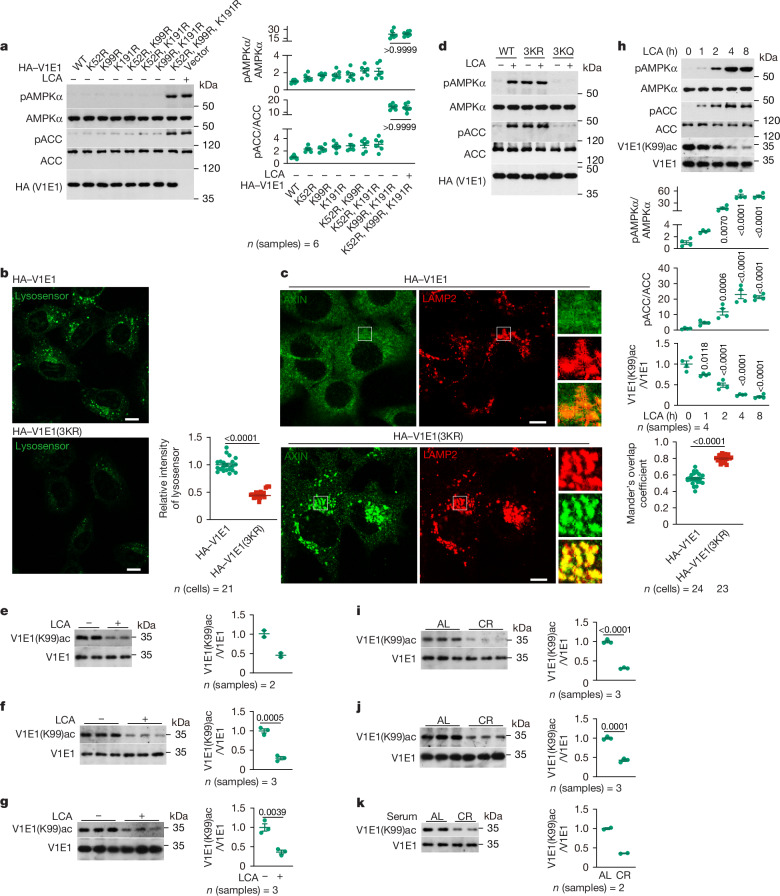
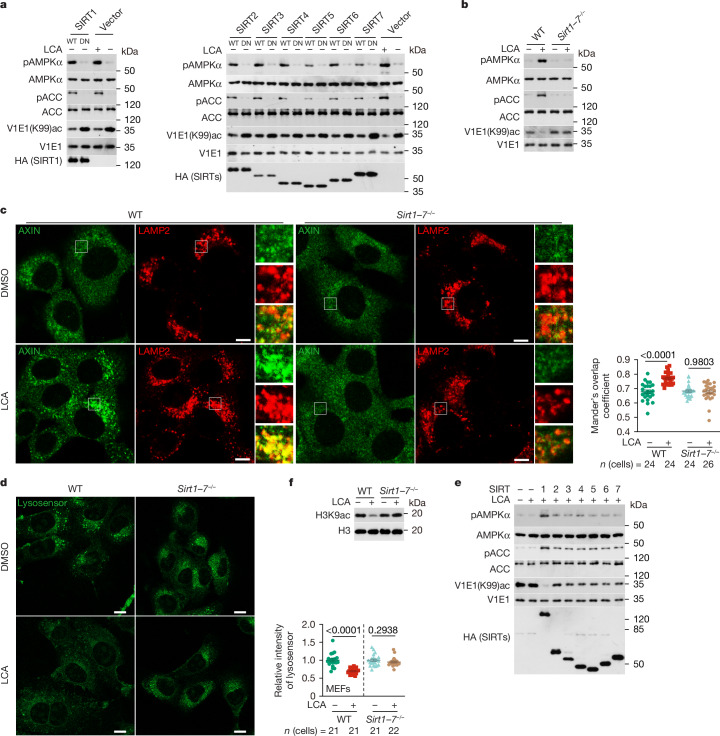
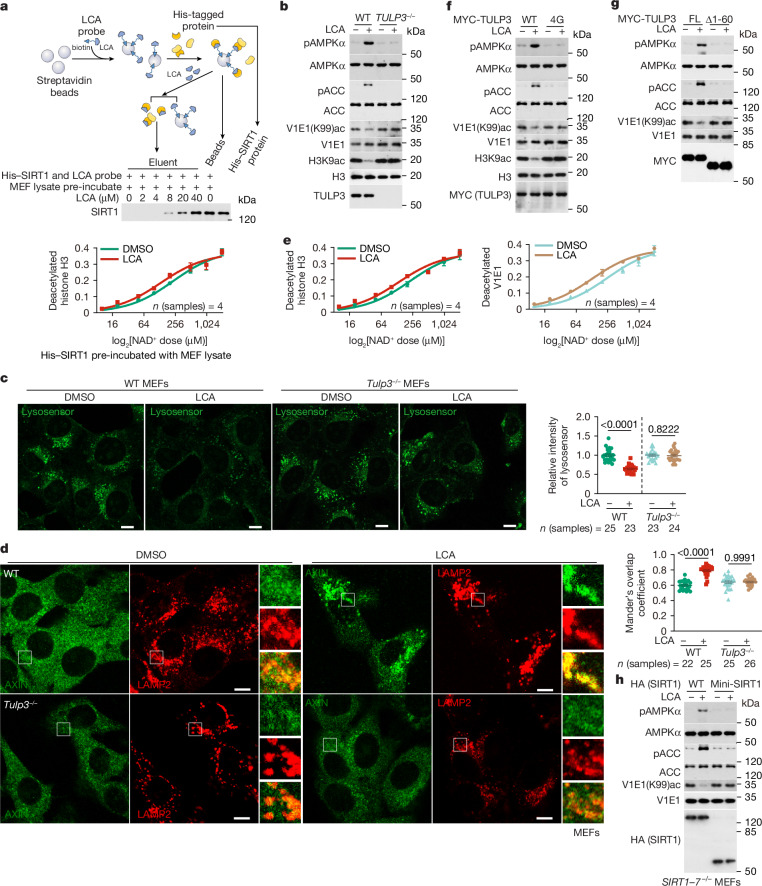
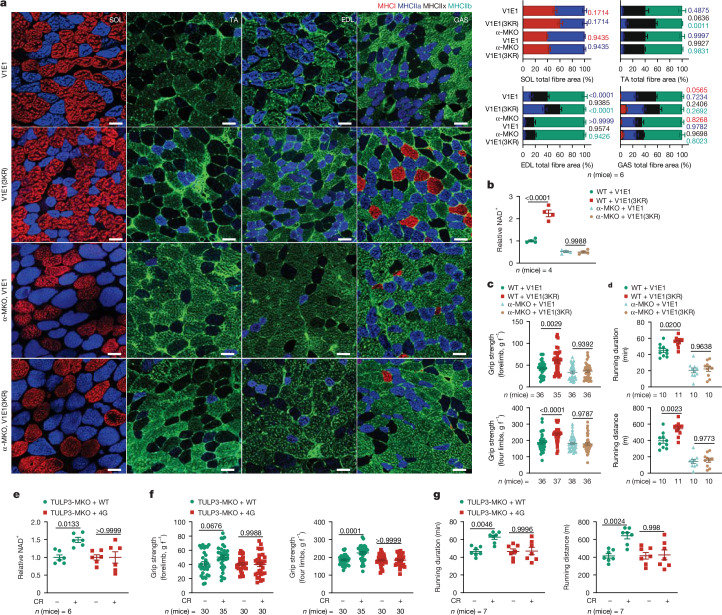
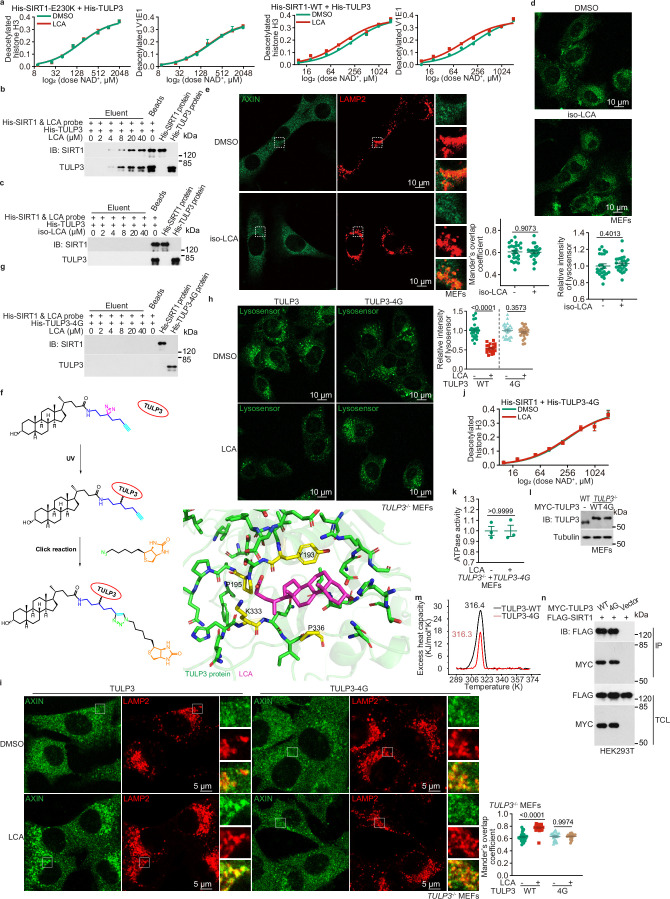
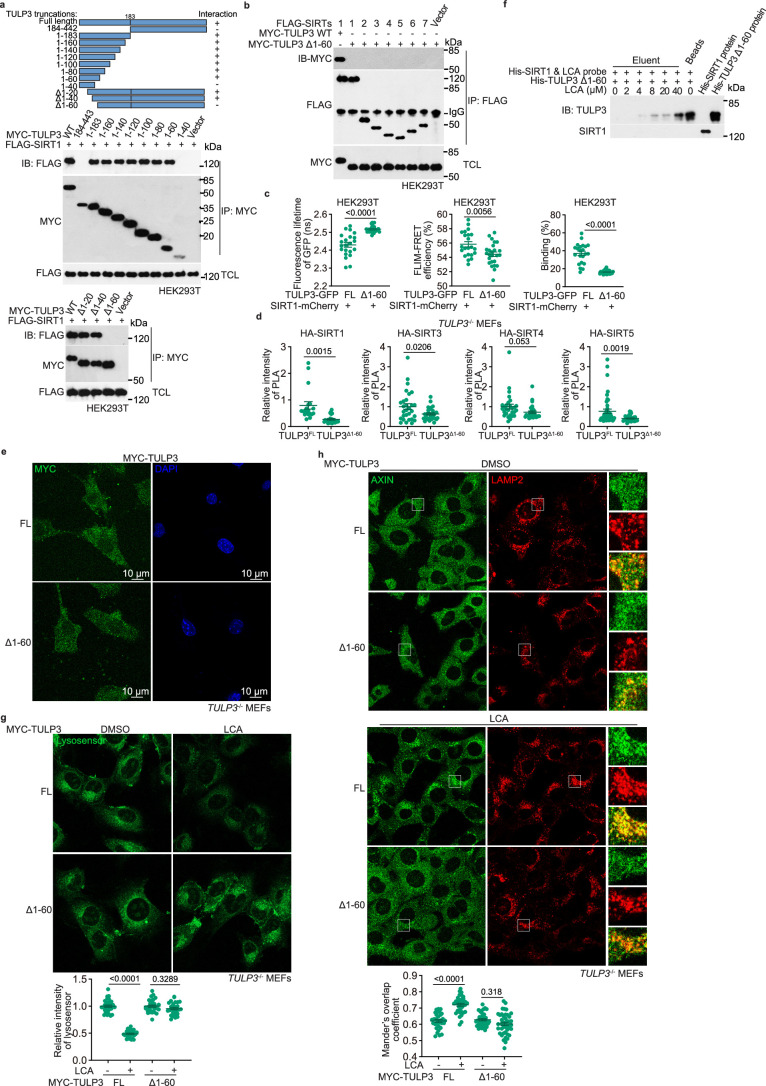
Lithocholic acid (LCA) is accumulated in mammals during calorie restriction and it can activate AMP-activated protein kinase (AMPK) to slow down ageing1. However, the molecular details of how LCA activates AMPK and induces these biological effects are unclear. Here we show that LCA enhances the activity of sirtuins to deacetylate and subsequently inhibit vacuolar H+-ATPase (v-ATPase), which leads to AMPK activation through the lysosomal glucose-sensing pathway. Proteomics analyses of proteins that co-immunoprecipitated with sirtuin 1 (SIRT1) identified TUB-like protein 3 (TULP3), a sirtuin-interacting protein2, as a LCA receptor. In detail, LCA-bound TULP3 allosterically activates sirtuins, which then deacetylate the V1E1 subunit of v-ATPase on residues K52, K99 and K191. Muscle-specific expression of a V1E1 mutant (3KR), which mimics the deacetylated state, strongly activates AMPK and rejuvenates muscles in aged mice. In nematodes and flies, LCA depends on the TULP3 homologues tub-1 and ktub, respectively, to activate AMPK and extend lifespan and healthspan. Our study demonstrates that activation of the TULP3-sirtuin-v-ATPase-AMPK pathway by LCA reproduces the benefits of calorie restriction.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
-
- Speakman, J. R. & Mitchell, S. E. Caloric restriction. Mol. Aspects Med.32, 159–221 (2011). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
