Diagnostic and prognostic implications of family history of fibrotic interstitial lung diseases
- PMID: 39695595
- PMCID: PMC11656921
- DOI: 10.1186/s12931-024-03063-y
Diagnostic and prognostic implications of family history of fibrotic interstitial lung diseases
Abstract
Background: Patients with familial fibrotic interstitial lung disease (ILD) experience worse survival than patients with sporadic disease. Current guidelines do not consider family aggregation or genetic information in the diagnostic algorithm for idiopathic pulmonary fibrosis or other fibrotic ILDs. Better characterizing familial cases could help in diagnostic and treatment decision-making.
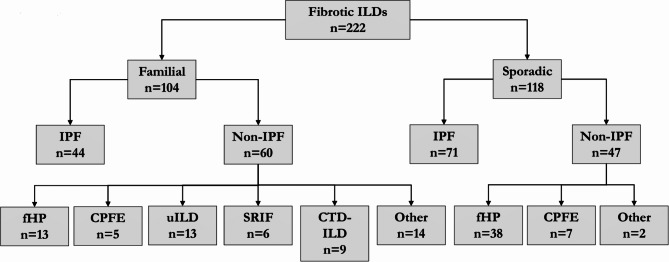
Methods: This retrospective cohort study included 222 patients with fibrotic ILD (104 familial and 118 sporadic) from Bellvitge University Hospital. Clinical, radiological, pulmonary functional tests (PFT), and histological evaluations were performed at diagnosis and follow-up. Telomere shortening and disease-associated variants (DAVs) in telomerase-related genes were analysed in familial patients and sporadic patients with telomeric clinical signs. Primary outcomes were the presence of a UIP histological pattern and disease progression.
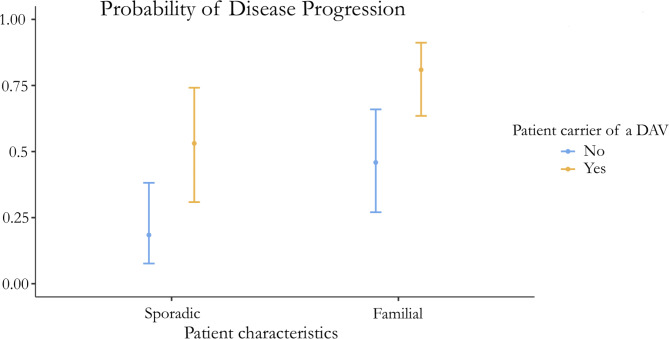
Results: Patients with idiopathic pulmonary fibrosis (IPF) (52%), fibrotic hypersensitivity pneumonitis (23%), and other fibrotic ILDs (25%) were included. 42% of patients underwent lung biopsy. Patients with family aggregation were younger and less frequently associated comorbidities, male sex, and smoking history. However, usual interstitial pneumonia (UIP) was more frequent on pathology (p = 0.005; OR 3.37), especially in patients with indeterminate or non-UIP radiological patterns. Despite similar PFT results at diagnosis, familial patients were more likely to present with progressive disease (p = 0.001; OR 3.75). Carrying a DAV increased the risk of fibrotic progression in familial and sporadic patients (p = 0.029, OR 5.01).
Discussion: Familial patients diagnosed with different fibrotic ILDs were more likely to exhibit a histological UIP pattern and disease progression than sporadic patients, independent of radiological findings and pulmonary function at diagnosis.
Conclusion: Considering the diagnostic likelihood of the histological UIP pattern and disease outcome, the presence of family aggregation would be useful in the decision making of multidisciplinary committees.
Keywords: Biopsy; Disease Progression; Family; Fibrosis; Idiopathic pulmonary fibrosis; Lung; Lung diseases, interstitial; Prognosis; Retrospective studies; Telomere Shortening.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The Ethics Committee of HUB approved the study (PR307/16), and all patients provided written informed consent before inclusion, including written informed consent for genetic testing if needed. Consent for publication: Not applicable. Clinical trial number: Not applicable. Competing interests: MMM has received grants for research purposes and fees for scientific advice outside this work from Boehringer Ing, Ferrer, Roche, Veracyte and Chiesi. The other authors of this work have no conflicts of interest to disclose.
Figures