

Genotypic and phenotypic analysis of an oculocutaneous albinism patient: a case report and review of the literature
- PMID: 39695711
- PMCID: PMC11654439
- DOI: 10.1186/s13256-024-04991-5
Genotypic and phenotypic analysis of an oculocutaneous albinism patient: a case report and review of the literature
Abstract
Background: Oculocutaneous albinism is a rare autosomal recessive disorder caused by congenital melanin deficiency, resulting in hypopigmentation of the eyes, hair, and skin. This study included a Chinese family with an oculocutaneous albinism pedigree, in which the proband presented with oculocutaneous albinismcombined with secondary angle closure, which has been rarely reported in previous literature. This article primarily focused on the clinical and genetic examination results of this patient and provided recommendations for ophthalmologist to treat patients with oculocutaneous albinism in clinical practice.
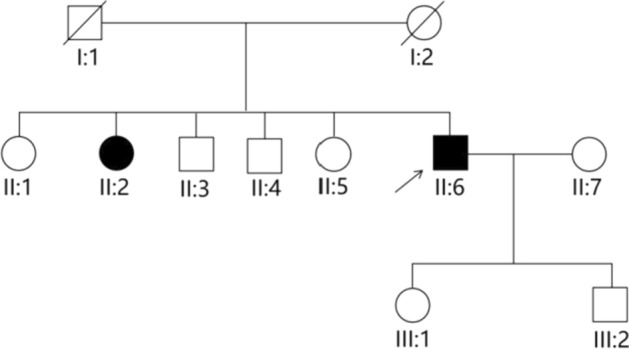
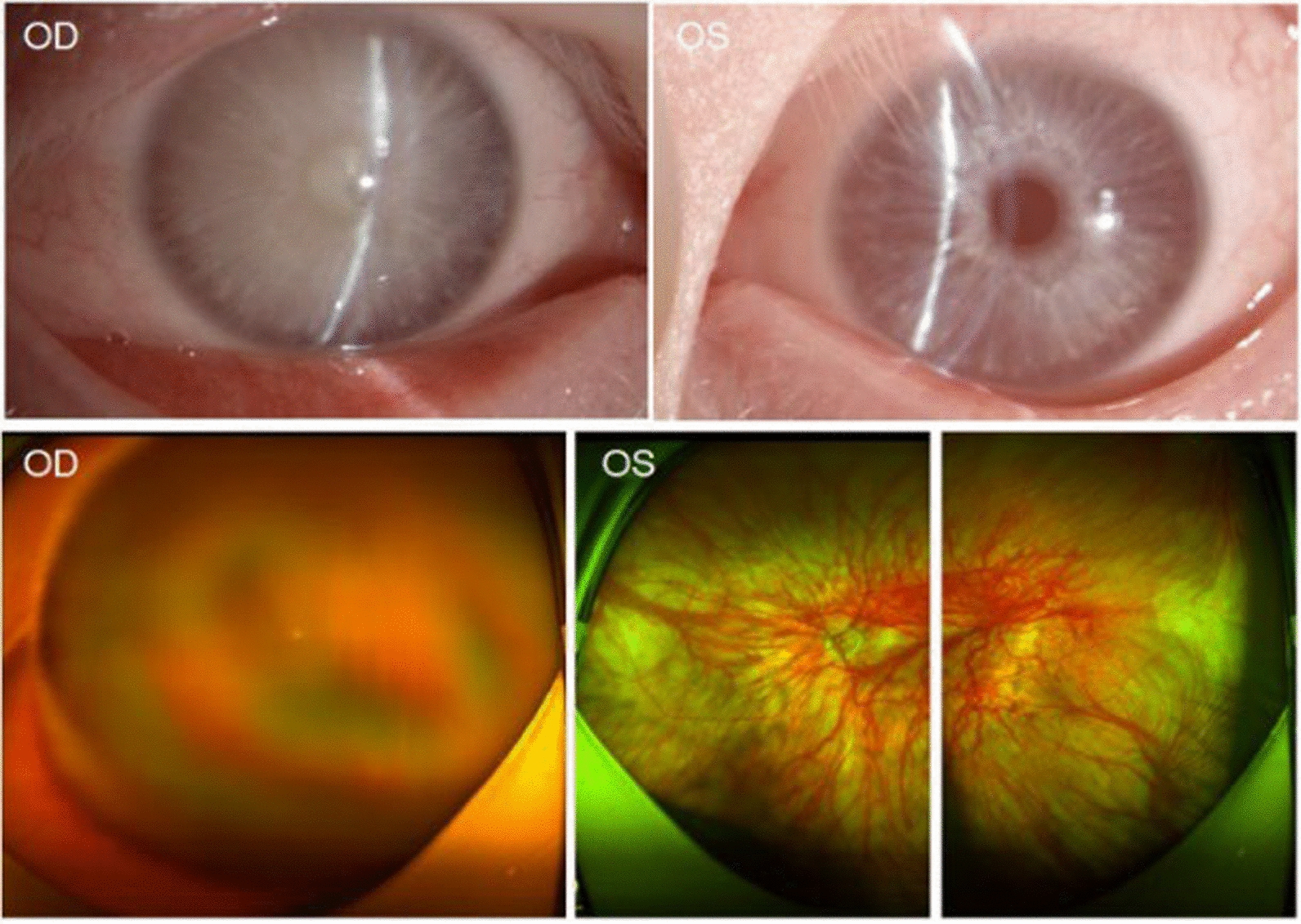
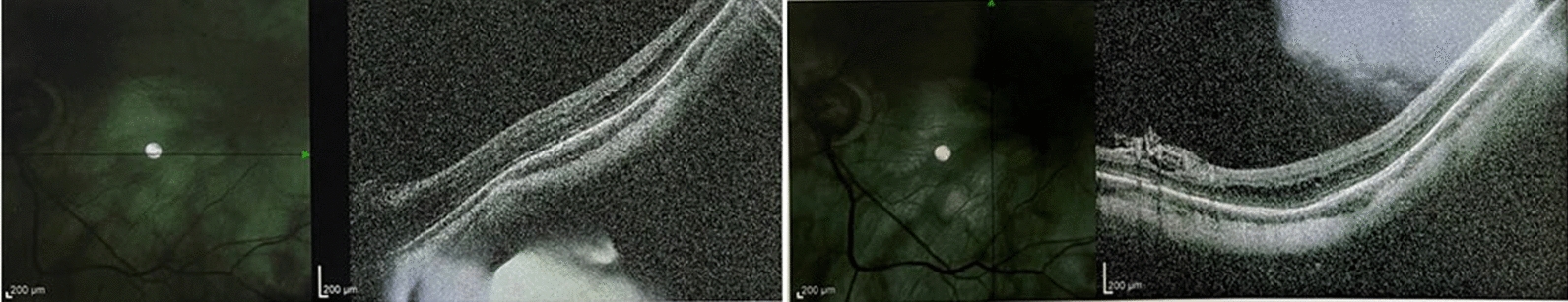
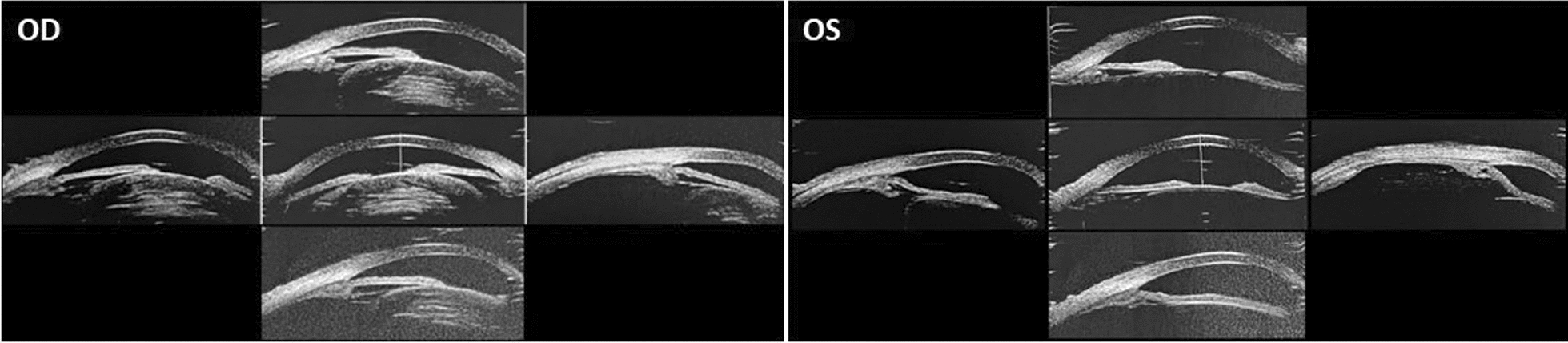
Case presentation: The proband in this case study is a 53-year-old Chinese male who showed depigmentation of the skin, hair, iris, and fundus, accompanied by photophobia, decreased vision, high intraocular pressure, nystagmus, macular fovea hypoplasia, and cataracts. Owing to the opacity and expansion of the lens, the volume ratio of lens to eyeball was increased, causing crowded anterior segment, bombed iris, and narrowed chamber angle and, ultimately, leading to secondary angle closure. Whole-exome sequencing suggested that the two patients in the pedigree harbored the compound heterozygous variants c.230G > A (p. Arg77Gln) and c.832G > A (p. Arg278*) in the TYR gene, while the healthy member carried the TYR c.230G > A (p. Arg77Gln) variant, which was consistent with the autosomal recessive inheritance pattern and further confirmed the diagnosis was oculocutaneous albinism. On the basis of the above results, the patient was diagnosed with oculocutaneous albinism, senile mature cataract, and secondary angle closure in the right eye and ocular hypertension in the left eye, as well as bilateral nystagmus. Then, the patient was prescribed carteolol eye drops to control intraocular pressure and underwent phacoemulsification and intraocular lens implantation surgery for the right eye. Postoperatively, the patient's intraocular pressure was effectively controlled, and visual acuity improved.
Conclusion: We report a patient with oculocutaneous albinism combined with cataract and secondary angle closure, and whole-exome sequencing suggested that he harbored TYR gene variants. Comprehensive examinations were important for identifying the causes of angle closure and making proper treatment strategies. Genetic testing enabled precise diagnosis and genetic counseling.
Keywords: TYR variant; Angle closure; Case report; Oculocutaneous albinism.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Ethical approval was not applicable. This study had no novel intervention and all treatments were standard treatments of an oculocutaneous albinism combined with angle closure. All patients gave written, informed consent to publish these cases and any accompanying images. Consent for publication: Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal. Competing interests: The authors declare that they have no competing interests.
Figures


Similar articles
-
Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism.Mol Vis. 2011 Apr 15;17:939-48. Mol Vis. 2011. PMID: 21541274 Free PMC article.
-
Genotype-phenotype associations in Danish patients with ocular and oculocutaneous albinism.Ophthalmic Genet. 2021 Jun;42(3):230-238. doi: 10.1080/13816810.2021.1881979. Epub 2021 Feb 22. Ophthalmic Genet. 2021. PMID: 33612058
-
Mild form of oculocutaneous albinism type 1: phenotypic analysis of compound heterozygous patients with the R402Q variant of the TYR gene.Br J Ophthalmol. 2019 Sep;103(9):1239-1247. doi: 10.1136/bjophthalmol-2018-312729. Epub 2018 Nov 24. Br J Ophthalmol. 2019. PMID: 30472657
-
Strabismus and nystagmus in oculocutaneous albinism: clinical perspectives, diagnosis, and role of neurotransmitters.Neurogenetics. 2025 Jun 18;26(1):50. doi: 10.1007/s10048-025-00830-x. Neurogenetics. 2025. PMID: 40531243 Review.
-
Oculocutaneous albinism type 1A: a case report.Dermatol Online J. 2008 Nov 15;14(11):13. Dermatol Online J. 2008. PMID: 19094851 Review.
References
-
- Khan J, Asif S, Ghani S, Khan H, Arshad MW, Khan SA, Lin S, Baple EL, Salter C, Crosby AH, Rawlins L, Shabbir MI. Mutational spectrum associated with oculocutaneous albinism and Hermansky-Pudlak syndrome in nine Pakistani families. BMC Ophthalmol. 2024;24(1):345. 10.1186/s12886-024-03611-6. - PMC - PubMed
-
- Seguy PH, Korobelnik JF, Delyfer MN, Michaud V, Arveiler B, Lasseaux E, Gattoussi S, Rougier MB, Trin K, Morice-Picard F, Ghomashchi N, Coste V. Ophthalmologic phenotype-genotype correlations in patients with oculocutaneous albinism followed in a reference center. Invest Ophthalmol Vis Sci. 2023;64(12):26. 10.1167/iovs.64.12.26. - PMC - PubMed
-
- Federico JR, Krishnamurthy K. Albinism. In: Federico JR, editor. StatPearls. Treasure Island: StatPearls Publishing; 2024. - PubMed
-
- Catalano RA, Nelson LB, Schaffer DB. Oculocutaneous albinism associated with congenital glaucoma. Ophthalmic Paediatr Genet. 1988;9(1):5–6. 10.3109/13816818809031473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
