

CGRclust: Chaos Game Representation for twin contrastive clustering of unlabelled DNA sequences
- PMID: 39695938
- PMCID: PMC11657719
- DOI: 10.1186/s12864-024-11135-y
CGRclust: Chaos Game Representation for twin contrastive clustering of unlabelled DNA sequences
Abstract
Background: Traditional supervised learning methods applied to DNA sequence taxonomic classification rely on the labor-intensive and time-consuming step of labelling the primary DNA sequences. Additionally, standard DNA classification/clustering methods involve time-intensive multiple sequence alignments, which impacts their applicability to large genomic datasets or distantly related organisms. These limitations indicate a need for robust, efficient, and scalable unsupervised DNA sequence clustering methods that do not depend on sequence labels or alignment.
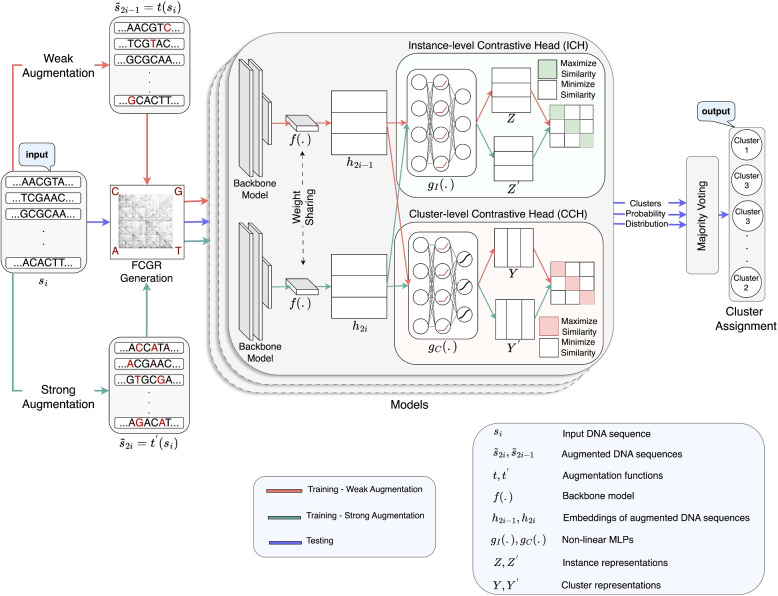
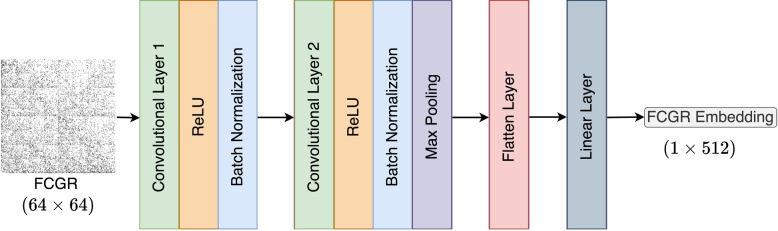
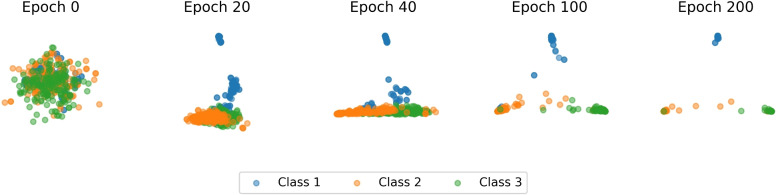
Results: This study proposes CGRclust, a novel combination of unsupervised twin contrastive clustering of Chaos Game Representations (CGR) of DNA sequences, with convolutional neural networks (CNNs). To the best of our knowledge, CGRclust is the first method to use unsupervised learning for image classification (herein applied to two-dimensional CGR images) for clustering datasets of DNA sequences. CGRclust overcomes the limitations of traditional sequence classification methods by leveraging unsupervised twin contrastive learning to detect distinctive sequence patterns, without requiring DNA sequence alignment or biological/taxonomic labels. CGRclust accurately clustered twenty-five diverse datasets, with sequence lengths ranging from 664 bp to 100 kbp, including mitochondrial genomes of fish, fungi, and protists, as well as viral whole genome assemblies and synthetic DNA sequences. Compared with three recent clustering methods for DNA sequences (DeLUCS, iDeLUCS, and MeShClust v3.0.), CGRclust is the only method that surpasses 81.70% accuracy across all four taxonomic levels tested for mitochondrial DNA genomes of fish. Moreover, CGRclust also consistently demonstrates superior performance across all the viral genomic datasets. The high clustering accuracy of CGRclust on these twenty-five datasets, which vary significantly in terms of sequence length, number of genomes, number of clusters, and level of taxonomy, demonstrates its robustness, scalability, and versatility.
Conclusion: CGRclust is a novel, scalable, alignment-free DNA sequence clustering method that uses CGR images of DNA sequences and CNNs for twin contrastive clustering of unlabelled primary DNA sequences, achieving superior or comparable accuracy and performance over current approaches. CGRclust demonstrated enhanced reliability, by consistently achieving over 80% accuracy in more than 90% of the datasets analyzed. In particular, CGRclust performed especially well in clustering viral DNA datasets, where it consistently outperformed all competing methods.
Keywords: Alignment-free DNA sequence comparison; Chaos Game Representation (CGR); Convolutional neural network; DNA sequence clustering; Data augmentation; Taxonomic classification; Twin contrastive learning; Unsupervised learning.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

References
-
- Lovich JE, Hart KM. Taxonomy: A history of controversy and uncertainty. Ecol Conserv Diamond-Backed Terrapin. 2018;37–50.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
