

Decoding the enigmatic estrogen paradox in pulmonary hypertension: delving into estrogen metabolites and metabolic enzymes
- PMID: 39695964
- PMCID: PMC11653592
- DOI: 10.1186/s11658-024-00671-w
Decoding the enigmatic estrogen paradox in pulmonary hypertension: delving into estrogen metabolites and metabolic enzymes
Abstract
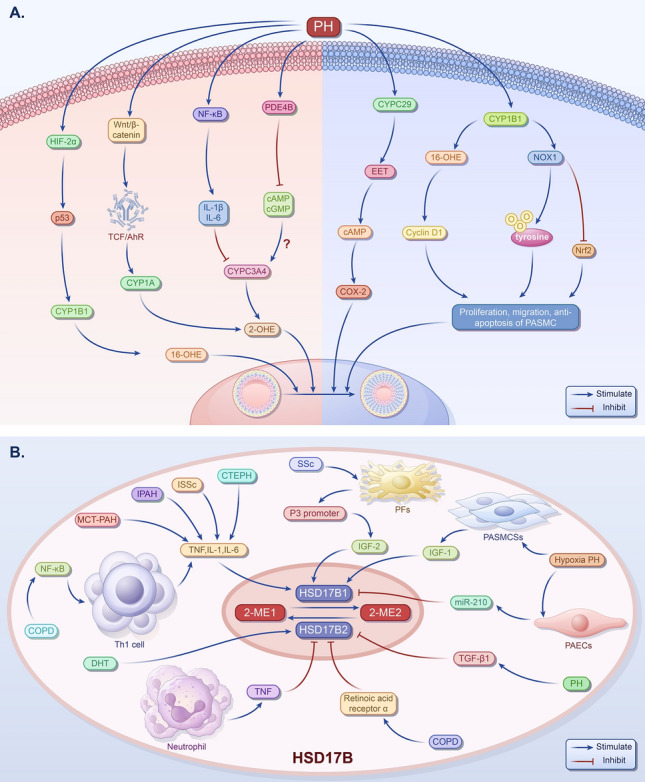
Pulmonary hypertension (PH) presents a puzzling sex bias, being more prevalent in women yet often less severe than in men, and the underlying reasons remain unclear. Studies using animal models, and limited clinical data have revealed a protective influence of exogenous estrogens, known as the estrogen paradox. Research suggests that beyond its receptor-mediated effects, estrogen acts through metabolites such as 2-ME2, 4-OHE2, and 16-OHE2, which are capable of exhibiting protective or detrimental effects in PH, prompting the need to explore their roles in PH to untangle sex differences and the estrogen paradox. Hypoxia disrupts the balance of estrogen metabolites by affecting the enzymes responsible for estrogen metabolism. Delving into the role of these metabolic enzymes not only illuminates the sex difference in PH but also provides a potential rationale for the estrogen paradox. This review delves into the intricate interplay between estrogen metabolites, metabolic enzymes, and PH, offering a deeper understanding of sex-specific differences and the perplexing estrogen paradox in the context of this condition.
Keywords: CYPs; Estrogen; Estrogen metabolites; HSD17B; Hypoxia; Pulmonary hypertension.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate.: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that there is no conflict of interest.
Figures
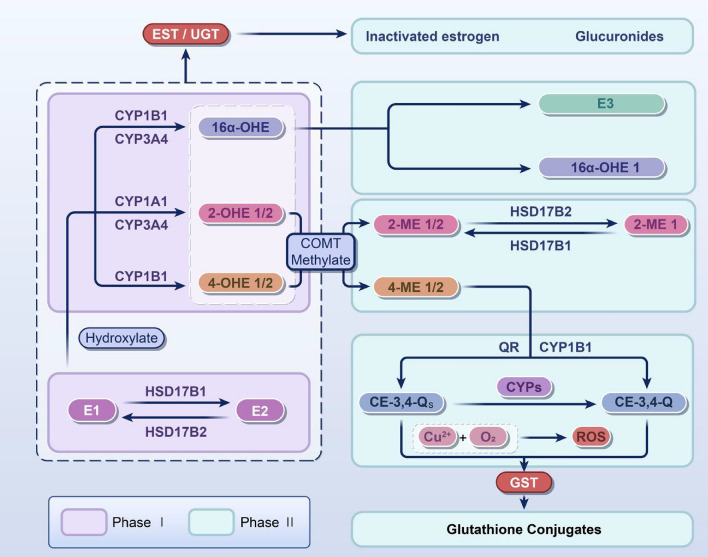
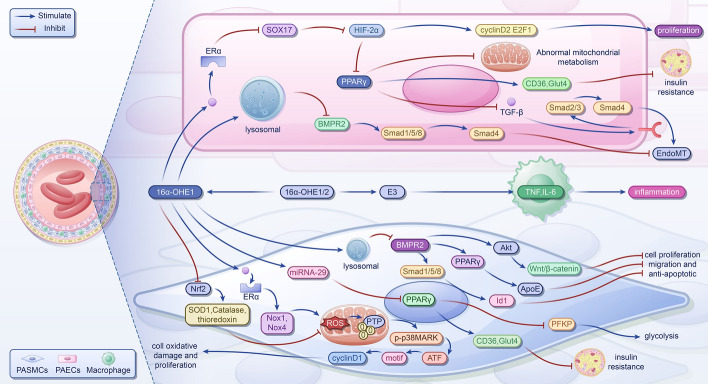
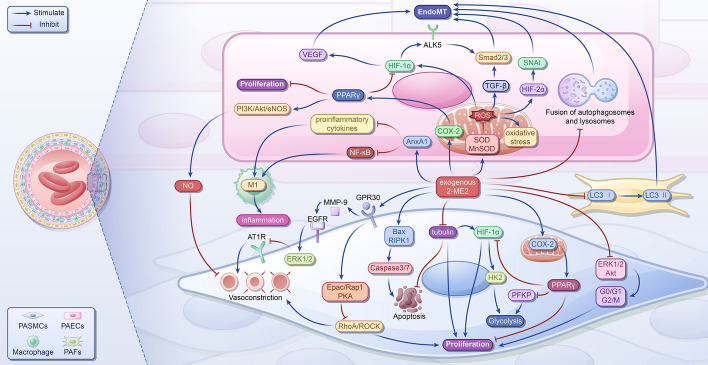
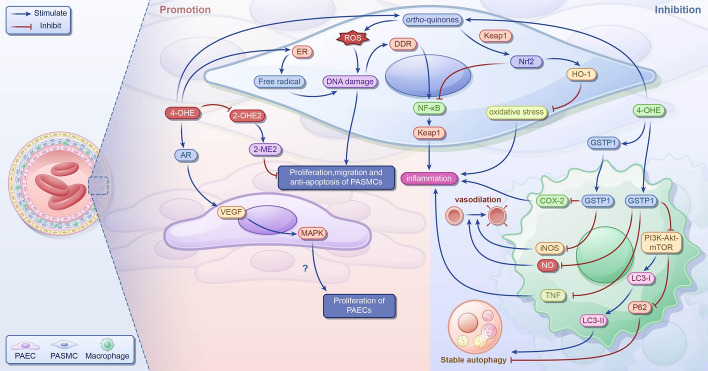
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
