

Evaluating data requirements for high-quality haplotype-resolved genomes for creating robust pangenome references
- PMID: 39696427
- PMCID: PMC11658127
- DOI: 10.1186/s13059-024-03452-y
Evaluating data requirements for high-quality haplotype-resolved genomes for creating robust pangenome references
Abstract
Background: Long-read technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) have transformed genomics research by providing diverse data types like HiFi, Duplex, and ultra-long ONT. Despite recent strides in achieving haplotype-phased gapless genome assemblies using long-read technologies, concerns persist regarding the representation of genetic diversity, prompting the development of pangenome references. However, pangenome studies face challenges related to data types, volumes, and cost considerations for each assembled genome, while striving to maintain sensitivity. The absence of comprehensive guidance on optimal data selection exacerbates these challenges.
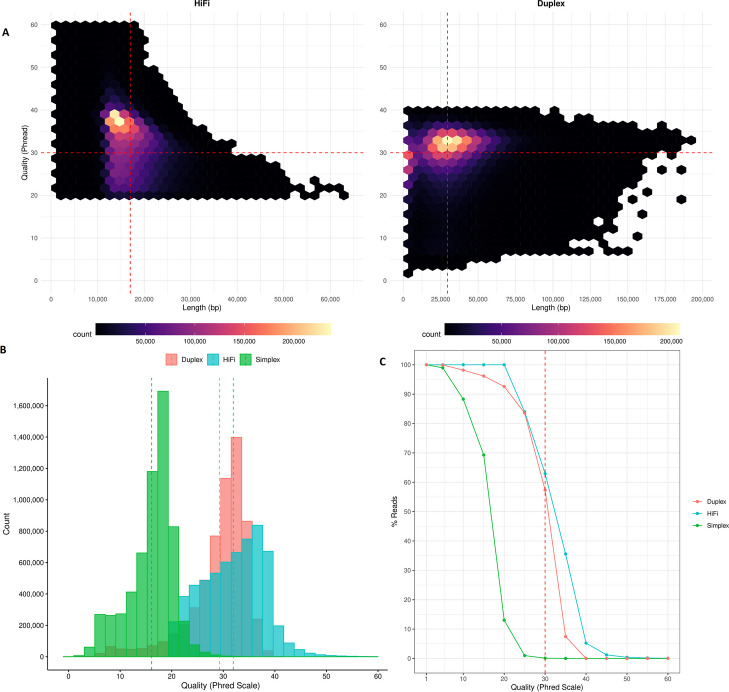
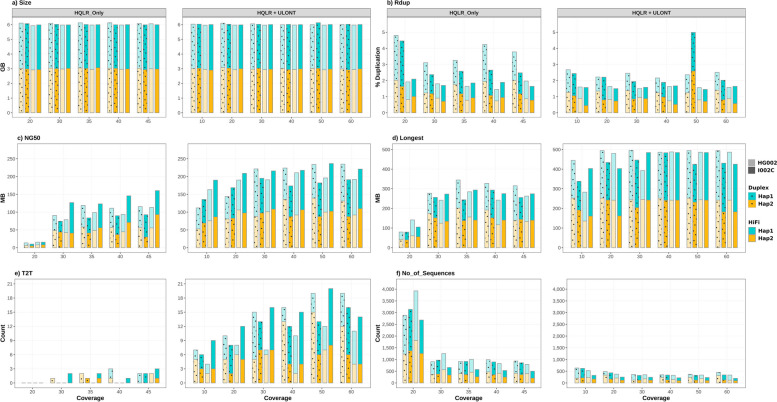
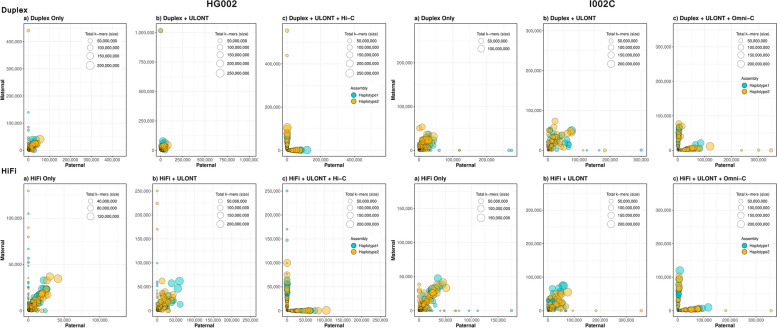
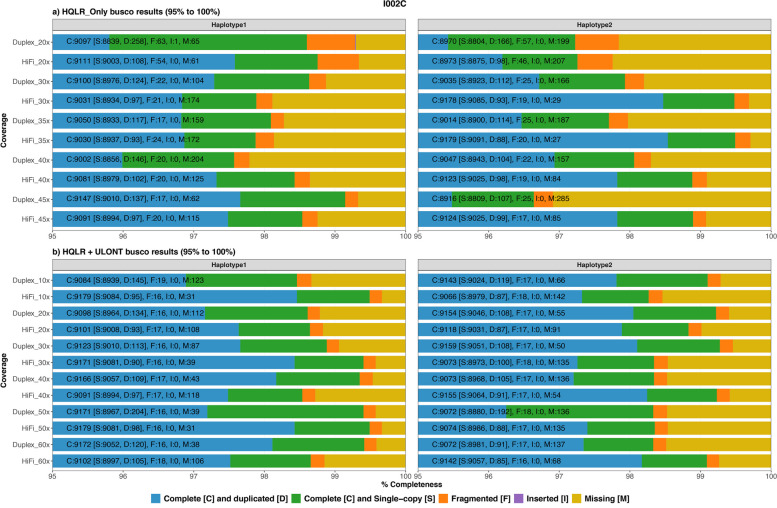
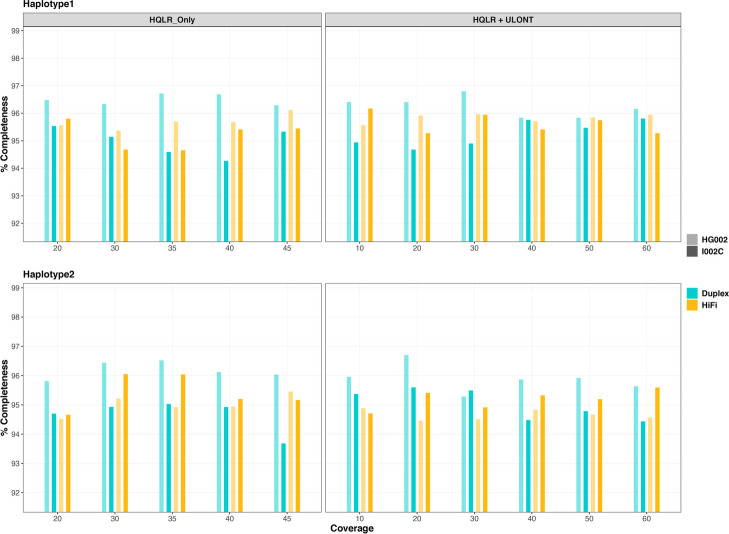
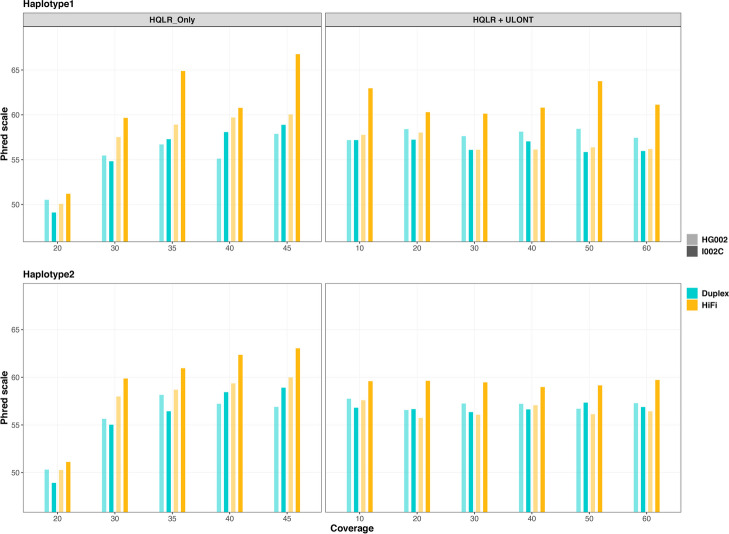
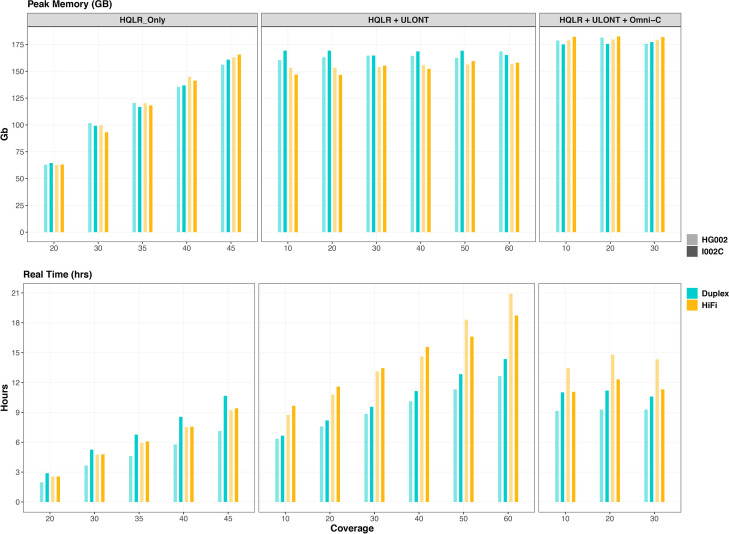
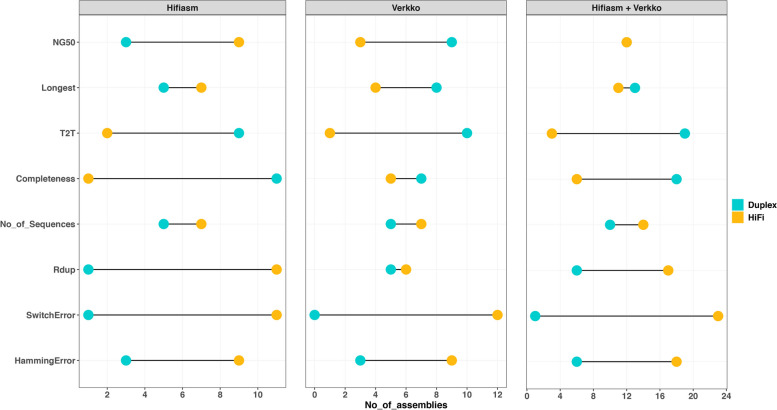
Results: Our study evaluates recommended data types and volumes required to establish a robust de novo genome assembly pipeline for population-level pangenome projects, extensively examining performance between ONT's Duplex and PacBio HiFi datasets in the context of achieving high-quality phased genomes with enhanced contiguity and completeness. The results show that achieving chromosome-level haplotype-resolved assembly requires 20 × high-quality long reads such as PacBio HiFi or ONT Duplex, combined with 15-20 × of ultra-long ONT per haplotype and 10 × of long-range data such as Omni-C or Hi-C. High-quality long reads from both platforms yield assemblies with comparable contiguity, with HiFi excelling in phasing accuracies, while Duplex generates more T2T contigs.
Conclusion: Our study provides insights into optimal data types and volumes for robust de novo genome assembly in population-level pangenome projects. Reassessing the recommended data types and volumes in this study and aligning them with practical economic limitations are vital to the pangenome research community, contributing to their efforts and pushing genomic studies with broader impacts.
Keywords: De novo assembly; LRS special issue; Pangenome; Population-level studies; Sequencing platforms.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All the participants were provided with informed consent for sample collection, and usage including making data publicly available via databases. Sample collection and usage were approved by SingHealth Centralised Institutional Review Board (IRB Reference: 2024–069). All experimental methods comply with the Helsinki Declaration. Consent for publication: Not applicable. Competing interests: M.Š. has been jointly funded by Oxford Nanopore Technologies and AI Singapore for the project AI-driven De Novo Diploid Assembler. The remaining authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
