

Clinical characteristics and genetic analysis of four pediatric patients with Kleefstra syndrome
- PMID: 39696517
- PMCID: PMC11657243
- DOI: 10.1186/s12920-024-02065-5
Clinical characteristics and genetic analysis of four pediatric patients with Kleefstra syndrome
Abstract
Background: Kleefstra syndrome spectrum (KLEFS) is an autosomal dominant disorder that can lead to intellectual disability and autism spectrum disorders. KLEFS encompasses Kleefstra syndrome-1 (KLEFS1) and Kleefstra syndrome-2 (KLEFS2), with KLEFS1 accounting for more than 75%. However, limited information is available regarding KLEFS2. KLEFS1 is caused by a subtelomeric chromosomal abnormality resulting in either deletion at the end of the long arm of chromosome 9, which contains the EHMT1 gene, or by variants in the EHMT1 gene and the KMT2C gene that cause KLEFS2.
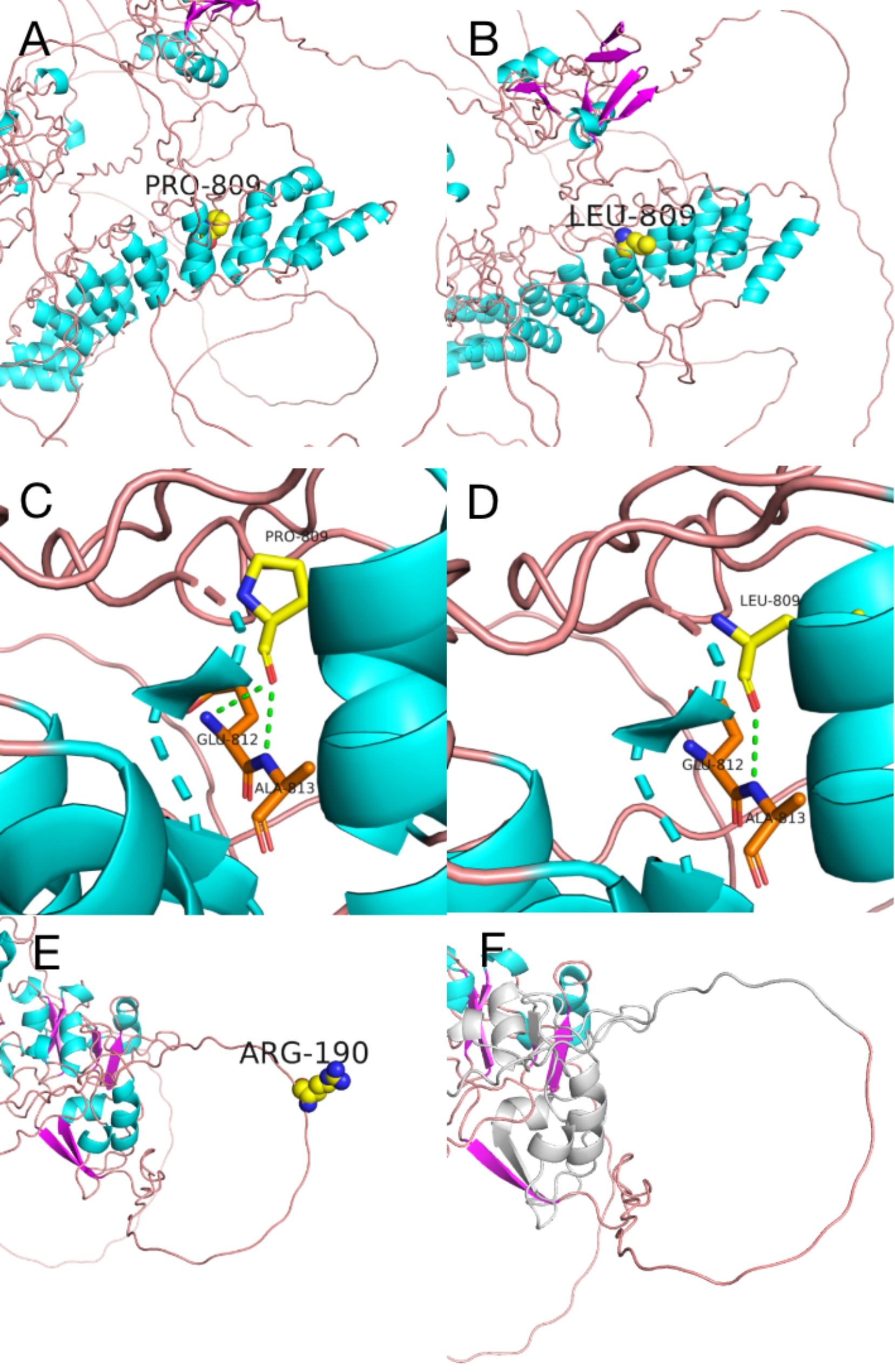
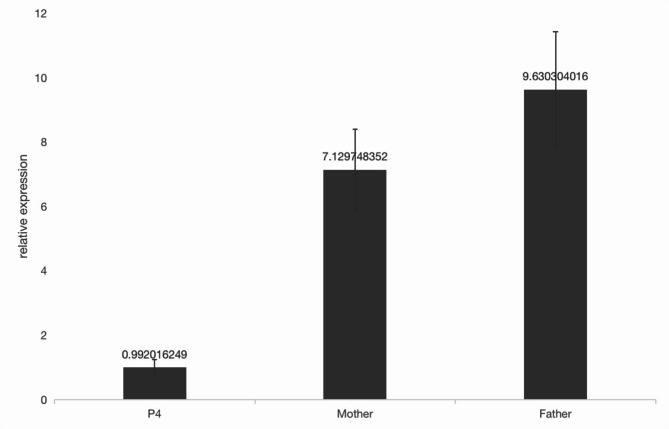
Methods: This study was a retrospective analysis of clinical data from four patients with KLEFS. Exome sequencing (ES) and Sanger sequencing techniques were used to identify and validate the candidate variants, facilitating the analysis of genotype‒phenotype correlations of the EHMT1 and KMT2C genes. Protein structure modeling was performed to evaluate the effects of the variants on the protein's three-dimensional structure. In addition, real-time quantitative reverse transcription‒polymerase chain reaction (RT‒qPCR) and western blotting were used to examine the protein and mRNA levels of the KMT2C gene.
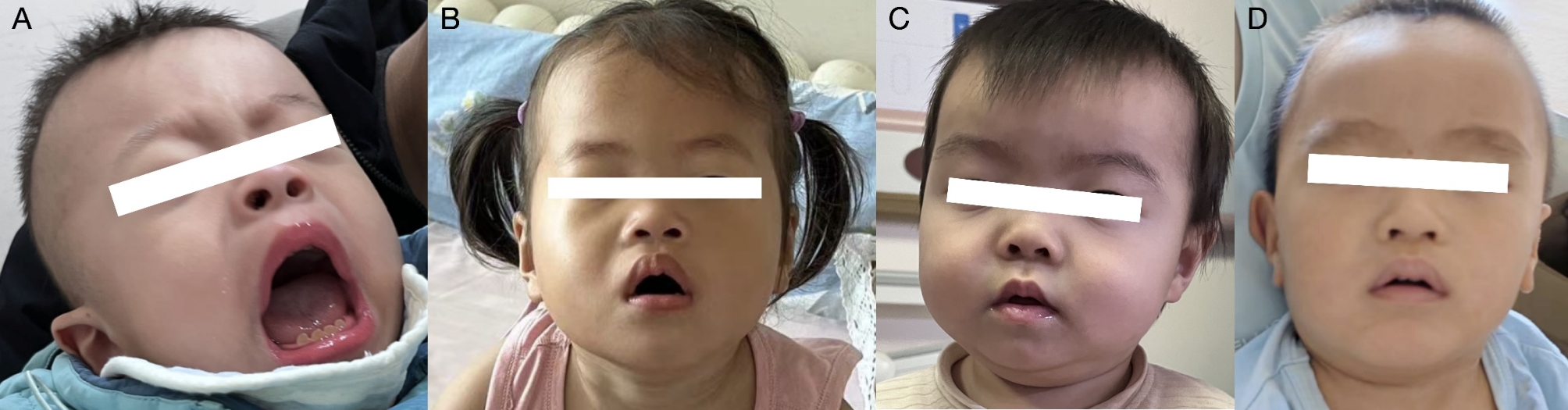
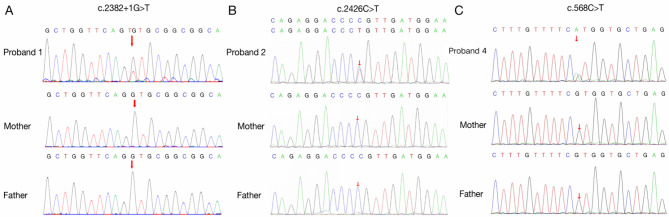
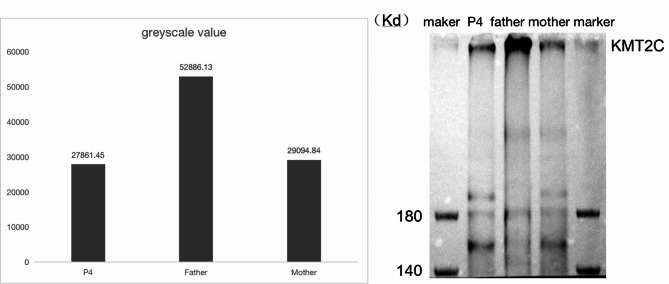
Results: Two patients with KLEFS1 were identified: one with a novel variant (c.2382 + 1G > T) and the other with a previously reported variant (c.2426 C > T, p.Pro809Leu) in the EHMT1 gene. A De novo deletion at the end of the long arm of chromosome 9 was also reported. Furthermore, a patient with KLEFS2 was identified with a novel variant in the KMT2C gene (c.568 C > T, p.Arg190Ter). The RT‒qPCR and western blot results revealed that the expression of the KMT2C gene was downregulated in the KLEFS2 sample.
Conclusion: This study contributes to the understanding of both KLEFS1 and KLEFS2 by identifying novel variants in EHMT1 and KMT2C genes, thereby expanding the variant spectrum. Additionally, we provide the first evidence of how a KMT2C variant leads to decreased gene and protein expression, enhancing our understanding of the molecular mechanisms underlying KLEFS2. Based on these findings, children exhibiting developmental delay, hypotonia, distinctive facial features, and other neurodevelopmental abnormalities should be considered for ES to ensure early intervention and treatment.
Keywords: EHMT1 gene; KMT2C gene; Exome sequencing (ES); Kleefstra syndrome-1 (KLEFS1); Kleefstra syndrome-2 (KLEFS2); RT‒qPCR.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of the Affiliated Women’s and Children’s Hospital of Qingdao University (QFELL-YJ-2024-06). Consent for publication: Informed consent was obtained from parents who consented to publish the images as well as the medical information supplied in this research. Competing interests: The authors declare no competing interests.
Figures
References
-
- Frega M, Selten M, Mossink B, Keller JM, Linda K, Moerschen R, et al. Distinct Pathogenic Genes Causing Intellectual Disability and Autism Exhibit a Common Neuronal Network Hyperactivity Phenotype. Cell Rep. 2020;30(1):173–e1866. - PubMed
-
- Guterman S, Hervé B, Rivière J, Fauvert D, Clement P, Vialard F. First prenatal diagnosis of a ‘pure’ 9q34.3 deletion (Kleefstra syndrome): A case report and literature review: Prenatal diagnosis of Kleefstra syndrome. J Obstet Gynaecol Res. 2018;44(3):570–5. - PubMed
-
- De Taevernier C, Meunier-Cussac S, Madigand J. First episode of psychosis in Kleefstra syndrome: a case report. Neurocase. 2021;27(3):227–30. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
