

Identification of the CD8+ T-cell exhaustion signature of hepatocellular carcinoma for the prediction of prognosis and immune microenvironment by integrated analysis of bulk- and single-cell RNA sequencing data
- PMID: 39697729
- PMCID: PMC11651828
- DOI: 10.21037/tcr-24-650
Identification of the CD8+ T-cell exhaustion signature of hepatocellular carcinoma for the prediction of prognosis and immune microenvironment by integrated analysis of bulk- and single-cell RNA sequencing data
Abstract
Background: Hepatocellular carcinoma (HCC) is a prevalent type of cancer with high incidence and mortality rates. It is the third most common cause of cancer-related deaths. CD8+ T cell exhaustion (TEX) is a progressive decline in T cell function due to sustained T cell receptor stimulation from continuous antigen exposure. Studies have shown that CD8+ TEX plays an important role in the anti-tumor immune process and is significantly correlated with patient prognosis. The aim of the research is to establish a reliable CD8+ TEX-based signature using single-cell RNA sequencing (scRNA-seq) and high-throughput RNA sequencing (RNA-seq), providing a new approach to evaluate HCC patient prognosis and immune microenvironment.
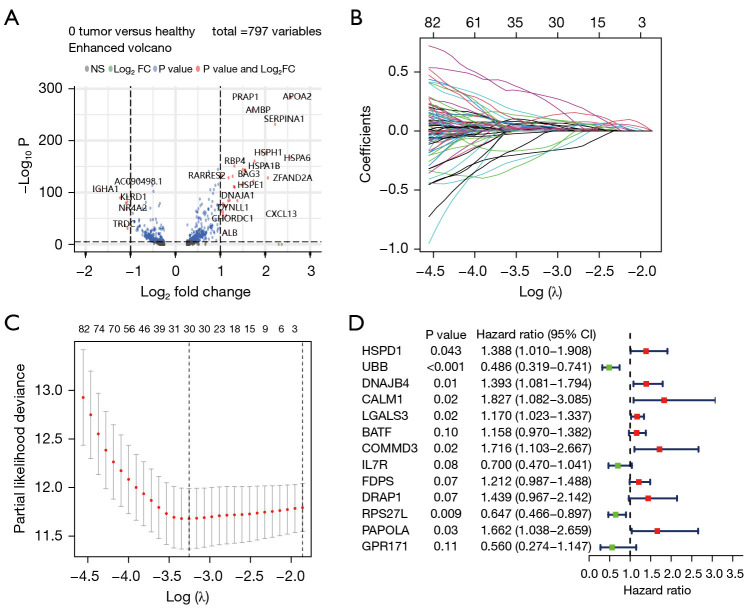
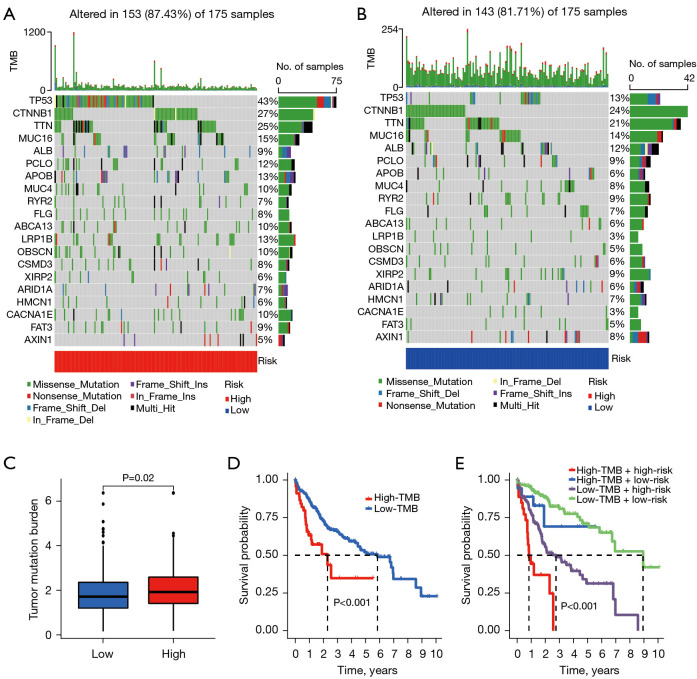
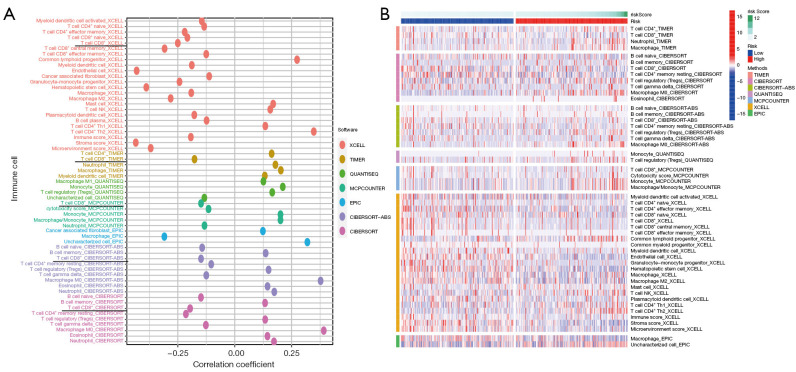
Methods: The RNA-seq data of HCC patients were download from three different databases: The Cancer Genome Atlas (TCGA), the Gene Expression Omnibus (GEO), and the International Cancer Genome Consortium (ICGC). HCC's 10× scRNA data were acquired from GSE149614. Based on single-cell sequencing data, CD8+ TEX-related genes were identified using uniform manifold approximation and projection (UMAP) algorithm, singleR, and marker gene methods. Afterwards, we proceeded to construct CD8+ TEX signature using differential gene analysis, univariate Cox regression analysis, least absolute shrinkage and selection operator (LASSO) regression, and multivariate Cox regression analysis. We also validated the CD8+ TEX signature in GEO and ICGC external cohorts and investigated clinical characteristics, chemotherapy sensitivity, mutation landscape, functional analysis, and immune cell infiltration in different risk groups.
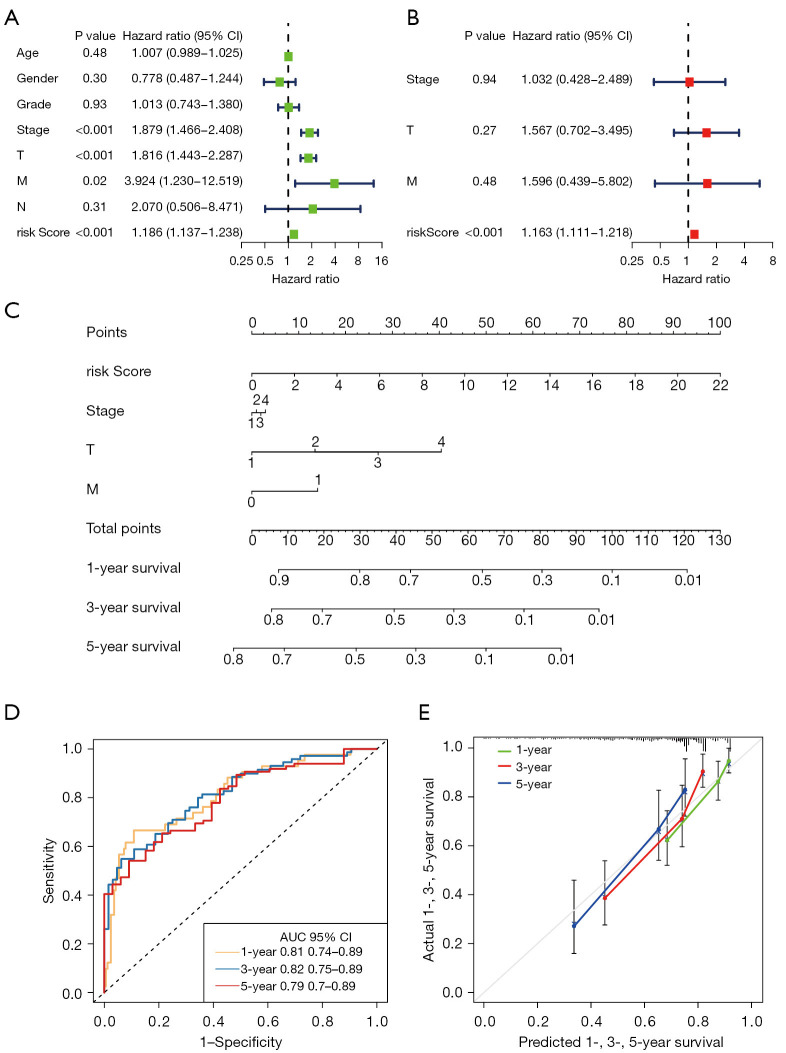
Results: The CD8+ TEX signature, consisting of 13 genes (HSPD1, UBB, DNAJB4, CALM1, LGALS3, BATF, COMMD3, IL7R, FDPS, DRAP1, RPS27L, PAPOLA, GPR171), was found to have a strong predictive effect on the prognosis of HCC. The Kaplan-Meier (KM) analysis showed that the overall survival (OS) rate of patients in the low-risk group was higher than that of patients in the high-risk group across different datasets and specific populations. The research findings suggested that the risk score was an independent predictor of HCC prognosis. The model based on clinical features and risk score has a strong predictive effect. We observed significant differences among various risk groups in terms of clinical characteristics, functional analysis, mutation landscape, chemotherapy sensitivity, and immune cell infiltration.
Conclusions: We constructed a CD8+ TEX signature to predict the survival probability of patients with HCC. We also found that the model could predict the sensitivity of targeted drugs and immune cell infiltration, and the risk score was negatively correlated with CD8+ T cell infiltration. In summary, the CD8+ TEX signature of HCC was constructed for the prediction of prognosis and immune microenvironment by integrated analysis of bulk and scRNA-seq data.
Keywords: CD8+ T cell exhaustion (CD8+ TEX); hepatocellular carcinoma (HCC); immunotherapy; prognosis; single-cell RNA sequencing (scRNA-seq).
2024 AME Publishing Company. All rights reserved.
Conflict of interest statement
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://tcr.amegroups.com/article/view/10.21037/tcr-24-650/coif). The authors have no conflicts of interest to declare.
Figures








References
-
- Shibutani M, Maeda K, Nagahara H, et al. The Prognostic Significance of the Tumor-infiltrating Programmed Cell Death-1(+) to CD8(+) Lymphocyte Ratio in Patients with Colorectal Cancer. Anticancer Res 2017;37:4165-72. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous