

Clinical Characterization of a National Cohort of Patients With Germline WT1 Variants Including Late-Onset Phenotypes
- PMID: 39698353
- PMCID: PMC11652072
- DOI: 10.1016/j.ekir.2024.09.007
Clinical Characterization of a National Cohort of Patients With Germline WT1 Variants Including Late-Onset Phenotypes
Abstract
Introduction: WT1 disorder is a recently introduced term for phenotypes associated with germline Wilms Tumor 1 (WT1) variants, including glomerulopathy, urogenital anomalies, and Wilms tumor. Previous studies showed a bias toward missense variants in the DNA-binding/Zinc-finger domain of WT1 (exon 8 and 9) and patients with early-onset glomerulopathy. Thorough genotype-phenotype correlations including follow-up data on late-onset glomerulopathy risk are lacking. To characterize the genotypic and phenotypic spectrum of WT1 disorder, we describe a national cohort of individuals with WT1 variants.
Methods: We requested clinical and genetic data of all patients with germline WT1 variants at all Dutch genetic laboratories.
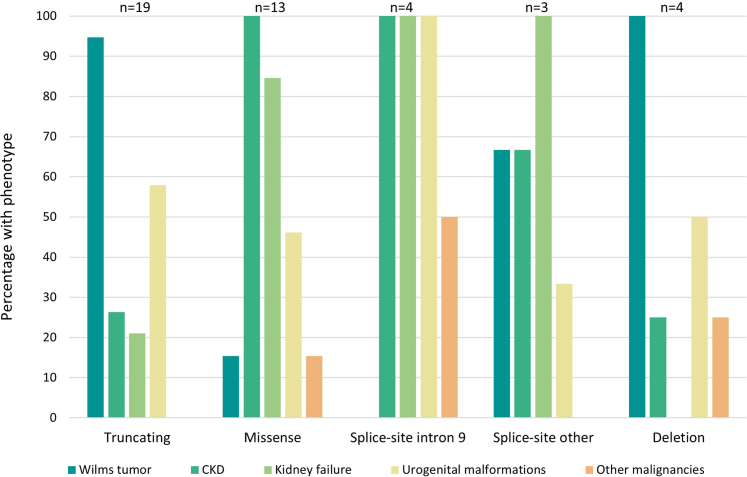
Results: We identified 43 patients with pathogenic WT1 variants (truncating, n = 19; missense, n = 13; splice-site, n = 7; and deletions, n = 4). Wilms tumor was the only clinical manifestation in 10 patients, of whom 9 were female. Wilms tumor occurred in 18 of 19 patients with truncating variants, in 4 of 4 patients with deletions, and was rarer in patients with missense or splice-site variants. All patients with missense and 6 of 7 with splice-site variants developed chronic kidney disease (CKD) versus 5 of 19 patients with truncating variants (3 in adulthood, with kidney failure at the age of 24, 26, and 41 years) and 1 of 4 with a deletion. Urogenital malformations occurred predominantly in 46,XY individuals.
Conclusion: Among patients with WT1 variants, a genotype-phenotype correlation was observed for Wilms tumor risk and age of CKD onset. Although childhood-onset CKD was more common in patients with missense variants in the DNA-binding/Zinc-finger domain, other patients may develop CKD and kidney failure later in life. Therefore, life-long surveillance of kidney function is recommended. Being alert about WT1 variants is especially important for girls with Wilms tumor who often miss additional phenotypes.
Keywords: Denys-Drash; WT1; Wilms tumor.
© 2024 International Society of Nephrology. Published by Elsevier Inc.
Figures



References
LinkOut - more resources
Full Text Sources
