

CFAP47 is Implicated in X-Linked Polycystic Kidney Disease
- PMID: 39698362
- PMCID: PMC11652189
- DOI: 10.1016/j.ekir.2024.09.013
CFAP47 is Implicated in X-Linked Polycystic Kidney Disease
Abstract
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is a well-described condition in which approximately 80% of all cases have a genetic explanation; and among sporadic cases without a family history, the genetic bases remain unclear in approximately 30% of cases. This study aimed to identify genes associated with polycystic kidney disease (PKD) in patients with sporadic cystic kidney disease in which a clear genetic change was not identified in established genes.
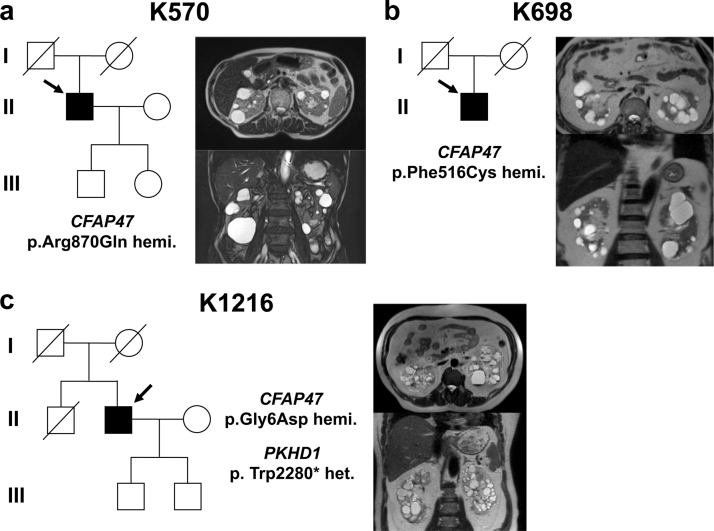
Methods: A next-generation sequencing panel analyzed known genes related to kidney cysts in 118 sporadic cases, followed by whole-genome sequencing (WGS) on 47 unrelated individuals without identified candidate variants. Immunohistology examination was then conducted on both human kidney tissue and kidneys from CFAP47-/Y mice.
Results: Three male patients were found to have rare missense variants in the X-linked gene cilia and flagella-associated protein 47 (CFAP47), none of whom had a family history of the condition. CFAP47 was expressed in primary cilia of human kidney tubules, and knockout (KO) mice exhibited vacuolation of tubular cells and tubular dilation, providing evidence that CFAP47 is a causative gene involved in cyst formation.
Conclusion: This discovery of CFAP47 as a newly identified gene associated with PKD, displaying X-linked inheritance, emphasizes the need for further cases to understand the role of CFAP47 in PKD.
Keywords: CFAP47; X-linked; polycystic kidney disease; sporadic.
© 2024 International Society of Nephrology. Published by Elsevier Inc.
Figures




Update of
-
CFAP47 is a novel causative gene implicated in X-linked polycystic kidney disease.medRxiv [Preprint]. 2024 Apr 5:2024.04.05.24304760. doi: 10.1101/2024.04.05.24304760. medRxiv. 2024. Update in: Kidney Int Rep. 2024 Sep 24;9(12):3580-3591. doi: 10.1016/j.ekir.2024.09.013. PMID: 38633811 Free PMC article. Updated. Preprint.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
