

An adenoviral vector encoding an inflammation-inducible antagonist, HMGB1 Box A, as a novel therapeutic approach to inflammatory diseases
- PMID: 39699172
- PMCID: PMC11796352
- DOI: 10.1128/mbio.03387-24
An adenoviral vector encoding an inflammation-inducible antagonist, HMGB1 Box A, as a novel therapeutic approach to inflammatory diseases
Abstract
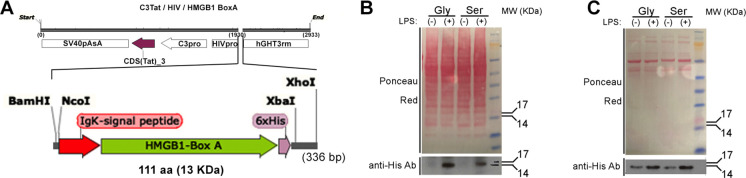
Influenza, as well as other respiratory viruses, can trigger local and systemic inflammation resulting in an overall "cytokine storm" that produces serious outcomes such as acute lung injury (ALI) or acute respiratory distress syndrome (ARDS). We hypothesized that gene therapy platforms could be useful in these cases if the production of an anti-inflammatory protein reflects the intensity and duration of the inflammatory condition. The recombinant protein would be produced and released only in the presence of the inciting stimulus, avoiding immunosuppression or other unwanted side effects that may occur when treating infectious diseases with anti-inflammatory drugs. To test this hypothesis, we developed AdV.C3-Tat/HIV-Box A, an inflammation-inducible cassette that remains innocuous in the absence of inflammation but releases HMGB1 Box A, an antagonist of high mobility group box 1 (HMGB1), in response to inflammatory stimuli such as lipopolysaccharide (LPS) or influenza virus infection. We report here that this novel inflammation-inducible HMGB1 Box A construct in a non-replicative adenovirus (AdV) vector mitigates lung and systemic inflammation therapeutically in response to influenza infection. We anticipate that this strategy will apply to the treatment of multiple diseases in which HMGB1-mediated signaling is a central driver of inflammation.IMPORTANCEMany inflammatory diseases are mediated by the action of a host-derived protein, HMGB1, on Toll-like receptor 4 (TLR4) to elicit an inflammatory response. We have engineered a non-replicative AdV vector that produces HMGB1 Box A, an antagonist of HMGB1-induced inflammation, under the control of an endogenous complement component C3 (C3) promoter sequence, that is inducible by LPS and influenza in vitro and ex vivo in macrophages (Mϕ) and protects mice and cotton rats therapeutically against infection with mouse-adapted and human non-adapted influenza strains, respectively, in vivo. We anticipate that this novel strategy will apply to the treatment of multiple infectious and non-infectious diseases in which HMGB1-mediated TLR4 signaling is a central driver of inflammation.
Keywords: Box A; HMGB1; LPS; MD-2; TLR4; adenovirus; cotton rats; influenza; mice.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
