

A Pan-RAS Inhibitor with a Unique Mechanism of Action Blocks Tumor Growth and Induces Antitumor Immunity in Gastrointestinal Cancer
- PMID: 39700396
- PMCID: PMC11875992
- DOI: 10.1158/0008-5472.CAN-24-0323
A Pan-RAS Inhibitor with a Unique Mechanism of Action Blocks Tumor Growth and Induces Antitumor Immunity in Gastrointestinal Cancer
Abstract
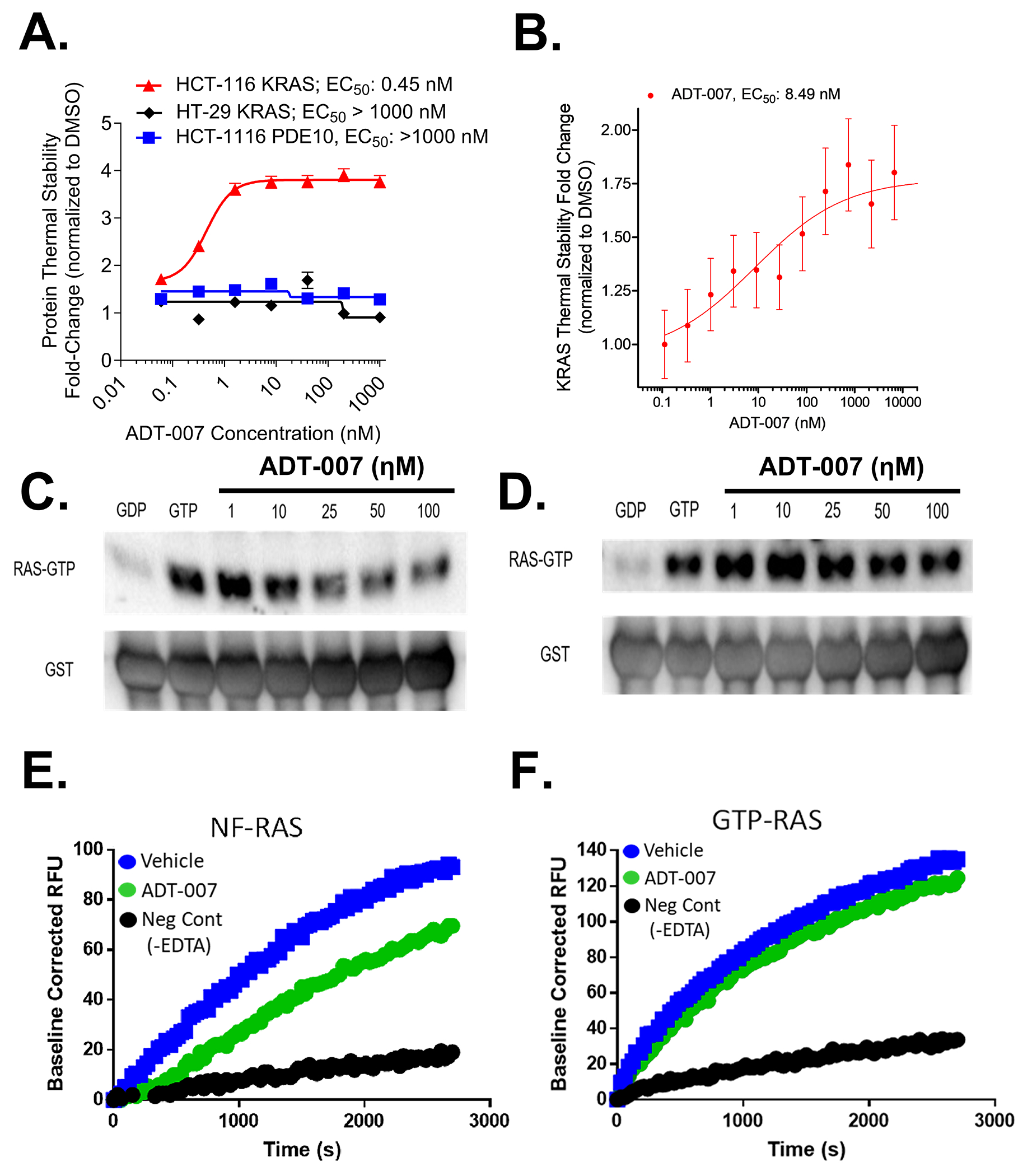
Activated RAS is a common driver of cancer that was considered undruggable for decades. Recent advances have enabled the development of RAS inhibitors, but the efficacy of these inhibitors remains limited by resistance. In this study, we developed a pan-RAS inhibitor, ADT-007, (Z)-2-(5-fluoro-1-(4-hydroxy-3,5-dimethoxybenzylidene)-2-methyl-1H-inden-3-yl)-N-(furan-2-ylmethyl)acetamide, that binds nucleotide-free RAS to block GTP activation of effector interactions and MAPK/AKT signaling, resulting in mitotic arrest and apoptosis. ADT-007 potently inhibited the growth of RAS-mutant cancer cells irrespective of the RAS mutation or isozyme. Wild-type RAS (RASWT) cancer cells with GTP-activated RAS from upstream mutations were equally sensitive. Conversely, RASWT cancer cells harboring downstream BRAF mutations and normal cells were essentially insensitive to ADT-007. Sensitivity of cancer cells to ADT-007 required activated RAS and dependence on RAS for proliferation, whereas insensitivity was attributed to metabolic deactivation by UDP-glucuronosyltransferases that were expressed in RASWT and normal cells but repressed in RAS-mutant cancer cells. ADT-007 displayed unique advantages over KRAS mutant-specific, pan-KRAS, and pan-RAS inhibitors that could impact in vivo antitumor efficacy by escaping compensatory mechanisms that lead to resistance. Local administration of ADT-007 showed robust antitumor activity in syngeneic immunocompetent and xenogeneic immune-deficient mouse models of colorectal and pancreatic cancers. The antitumor activity of ADT-007 was associated with the suppression of MAPK signaling and activation of innate and adaptive immunity in the tumor immune microenvironment. Oral administration of ADT-007 prodrug also inhibited tumor growth. Thus, ADT-007 has the potential to address the complex RAS mutational landscape of many human cancers and to improve treatment of RAS-driven tumors. Significance: ADT-007, a first-in-class pan-RAS inhibitor, has unique selectivity for cancer cells with mutant RAS or activated RAS protein and the capability to circumvent resistance to suppress tumor growth, supporting further development of ADT-007 analogs.
©2025 American Association for Cancer Research.
Conflict of interest statement
Figures





Update of
-
A Novel Pan-RAS Inhibitor with a Unique Mechanism of Action Blocks Tumor Growth in Mouse Models of GI Cancer.bioRxiv [Preprint]. 2024 Oct 7:2023.05.17.541233. doi: 10.1101/2023.05.17.541233. bioRxiv. 2024. Update in: Cancer Res. 2025 Mar 03;85(5):956-972. doi: 10.1158/0008-5472.CAN-24-0323. PMID: 38328254 Free PMC article. Updated. Preprint.
References
-
- Siegel RL, Giaquinto AN, Jemal A: Cancer statistics, 2024. CA Cancer J Clin 2024, 74(1):12–49. - PubMed
-
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4(6):437–450. - PubMed
-
- Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, Grizzle WE, Klug CA: Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res 2007, 67(17):8121–8130. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
