

Case report: A single novel calpain 3 gene variant associated with mild myopathy
- PMID: 39703226
- PMCID: PMC11655484
- DOI: 10.3389/fgene.2024.1437859
Case report: A single novel calpain 3 gene variant associated with mild myopathy
Abstract
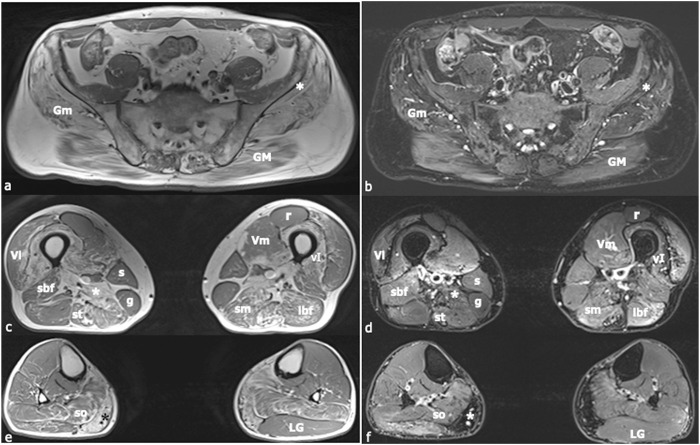
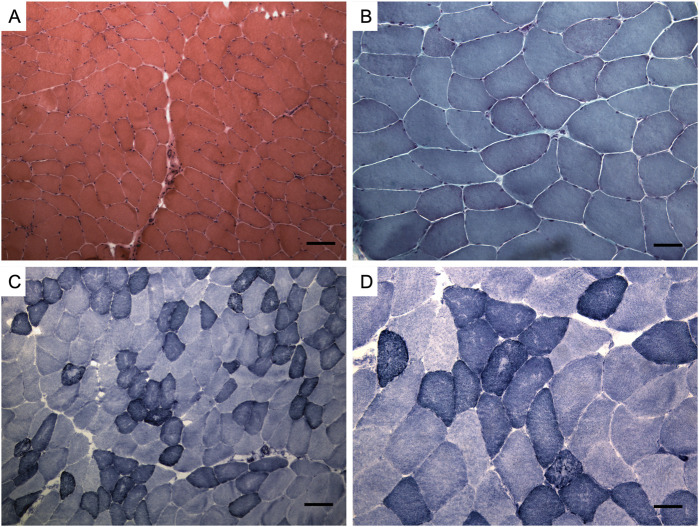
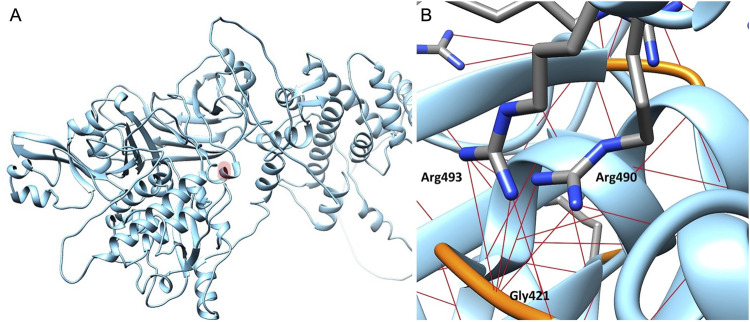
Recessively inherited limb-girdle muscular dystrophy type 1, caused by mutations in the calpain 3 gene, is the most common limb-girdle muscular dystrophy worldwide. Recently, cases of autosomal dominant calpainopathy have been described. A man was referred to our neurological outpatient clinic at the age of 54 for persistent hyperCKemia (>1000 U/l) associated with muscle fatigue and myalgia. Clinical examination revealed mild proximal weakness in the lower limbs. His brother exhibited a moderate increase in serum creatine kinase levels (up to 2000 U/l) without other signs of myopathy. Their father experienced slowly progressive lower limb weakness after the age of 50. The calpain 3 variant c.1478G>A (p.Arg493Gln) in the heterozygous state was identified in both brothers. In silico modeling studies predict that this substitution may disrupt protein folding. This represents the first description of the heterozygous p.Arg493Gln calpain 3 variant as a potential cause of mild calpainopathy.
Keywords: LGMD; LGMDR1; calpain; calpainopathy; case report.
Copyright © 2024 Massucco, Fossa, Fiorillo, Faedo, Gemelli, Barresi, Ripolone, Patrone, Gaudio, Mandich, Gotta, Baratto, Traverso, Pisciotta, Zaottini, Camera, Scarsi and Grandis.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources