Severe Adult-Onset Non-Dystrophic Myotonia With Apnea and Laryngospasm Due to Digenic Inheritance of SCN4A and CLCN1 Variants: A Case Report
- PMID: 39703462
- PMCID: PMC11655165
- DOI: 10.1212/NXG.0000000000200223
Severe Adult-Onset Non-Dystrophic Myotonia With Apnea and Laryngospasm Due to Digenic Inheritance of SCN4A and CLCN1 Variants: A Case Report
Abstract
Objectives: To report a case of adult-onset non-dystrophic myotonia complicated by recurrent episodes of laryngospasm.
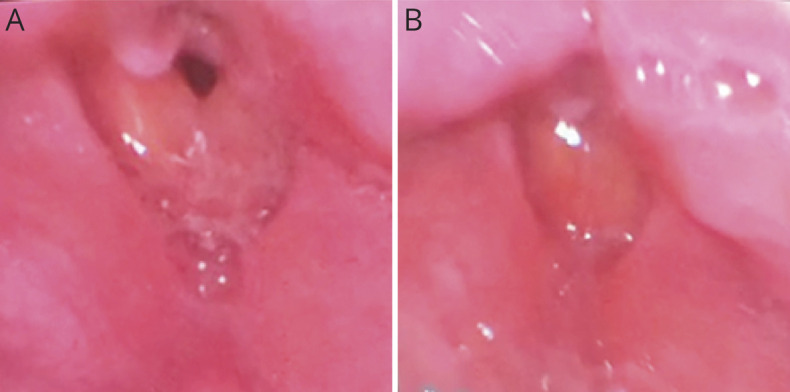
Methods: The patient is a 35-year-old man who was admitted to our hospital for recurrent episodes of apnea requiring endotracheal intubation with mechanical ventilation. He underwent extensive evaluation, including EMG, laryngoscopy, muscle biopsy, and genetic testing, which revealed a diagnosis of non-dystrophic myotonia.
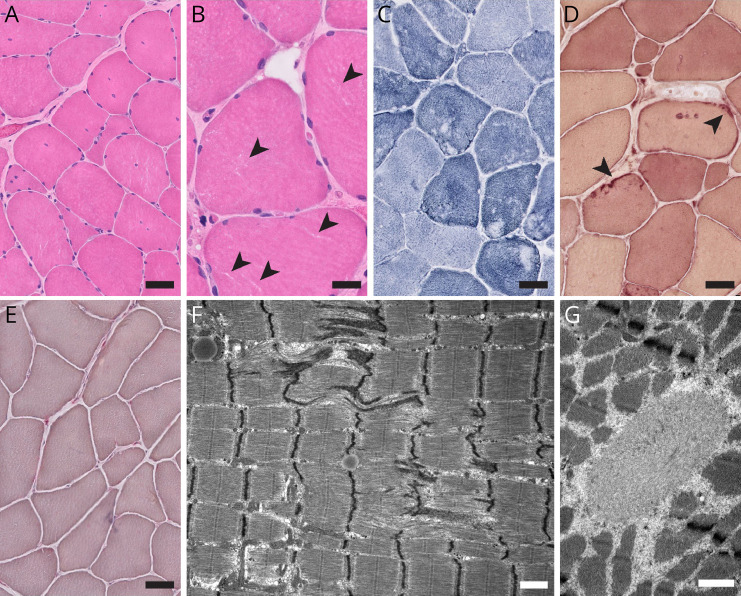
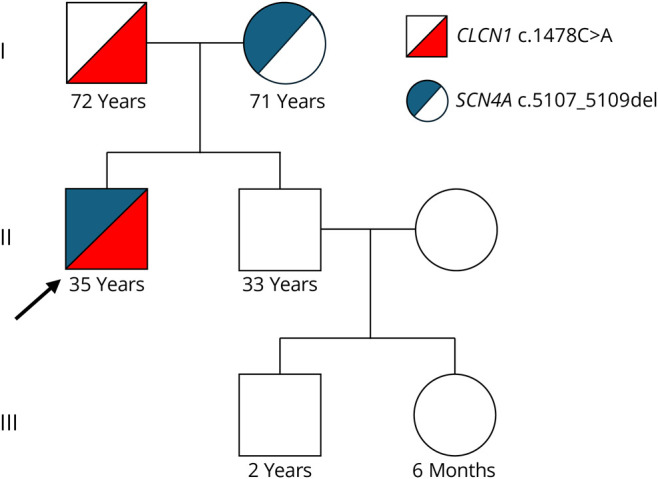
Results: His myotonic disorder was due to the synergistic effects of a pathogenic CLCN1 variant and a newly reported SCN4A variant. His muscle biopsy demonstrated myofibrillar disorganization with Z-band streaming, which may reflect the severity of his clinical and electrographic myotonia. Treatment with mexiletine resulted in resolution of his episodes of laryngospasm and symptoms of myotonia in the extremities.
Discussion: Our case adds to the literature on the potentiating effects of chloride channelopathies on sodium channel myotonia. This is the first reported case of an adult-onset sodium channelopathy manifesting with respiratory failure due to laryngospasm. In addition, we present muscle biopsy findings that have not been described in the recent literature. This case also highlights that a myotonic disorder should be considered in the differential diagnosis for recurrent episodes of mixed hypoxic and hypercarbic respiratory failure.
Copyright © 2024 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Conflict of interest statement
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
Figures