

Mitochondrial pathways of copper neurotoxicity: focus on mitochondrial dynamics and mitophagy
- PMID: 39703721
- PMCID: PMC11655512
- DOI: 10.3389/fnmol.2024.1504802
Mitochondrial pathways of copper neurotoxicity: focus on mitochondrial dynamics and mitophagy
Abstract
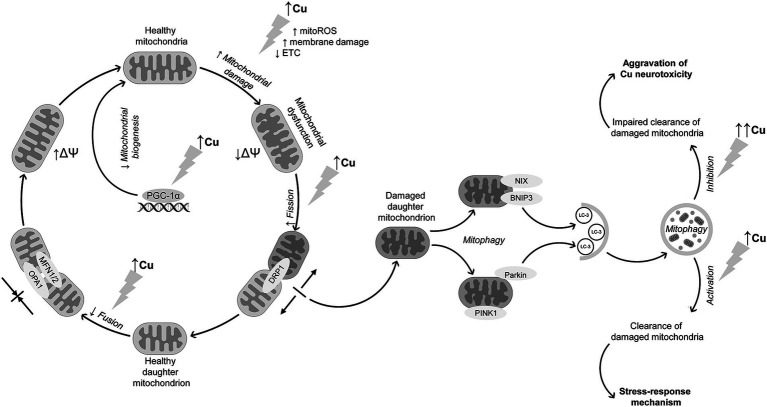
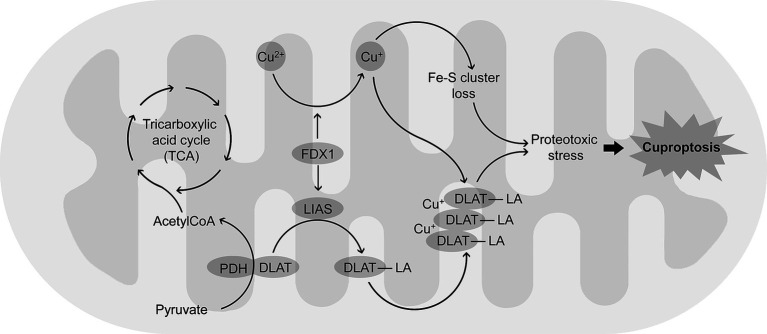
Copper (Cu) is essential for brain development and function, yet its overload induces neuronal damage and contributes to neurodegeneration and other neurological disorders. Multiple studies demonstrated that Cu neurotoxicity is associated with mitochondrial dysfunction, routinely assessed by reduction of mitochondrial membrane potential. Nonetheless, the role of alterations of mitochondrial dynamics in brain mitochondrial dysfunction induced by Cu exposure is still debatable. Therefore, the objective of the present narrative review was to discuss the role of mitochondrial dysfunction in Cu-induced neurotoxicity with special emphasis on its influence on brain mitochondrial fusion and fission, as well as mitochondrial clearance by mitophagy. Existing data demonstrate that, in addition to mitochondrial electron transport chain inhibition, membrane damage, and mitochondrial reactive oxygen species (ROS) overproduction, Cu overexposure inhibits mitochondrial fusion by down-regulation of Opa1, Mfn1, and Mfn2 expression, while promoting mitochondrial fission through up-regulation of Drp1. It has been also demonstrated that Cu exposure induces PINK1/Parkin-dependent mitophagy in brain cells, that is considered a compensatory response to Cu-induced mitochondrial dysfunction. However, long-term high-dose Cu exposure impairs mitophagy, resulting in accumulation of dysfunctional mitochondria. Cu-induced inhibition of mitochondrial biogenesis due to down-regulation of PGC-1α further aggravates mitochondrial dysfunction in brain. Studies from non-brain cells corroborate these findings, also offering additional evidence that dysregulation of mitochondrial dynamics and mitophagy may be involved in Cu-induced damage in brain. Finally, Cu exposure induces cuproptosis in brain cells due mitochondrial proteotoxic stress, that may also contribute to neuronal damage and pathogenesis of certain brain diseases. Based on these findings, it is assumed that development of mitoprotective agents, specifically targeting mechanisms of mitochondrial quality control, would be useful for prevention of neurotoxic effects of Cu overload.
Keywords: copper; cuproptosis; fission; mitochondrial fusion; mitophagy.
Copyright © 2024 Aschner, Skalny, Lu, Martins, Tizabi, Nekhoroshev, Santamaria, Sinitskiy and Tinkov.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Mutual Antagonism of PINK1/Parkin and PGC-1α Contributes to Maintenance of Mitochondrial Homeostasis in Rotenone-Induced Neurotoxicity.Neurotox Res. 2019 Feb;35(2):331-343. doi: 10.1007/s12640-018-9957-4. Epub 2018 Sep 21. Neurotox Res. 2019. PMID: 30242625
-
Induction of mitophagy via ROS-dependent pathway protects copper-induced hypothalamic nerve cell injury.Food Chem Toxicol. 2023 Nov;181:114097. doi: 10.1016/j.fct.2023.114097. Epub 2023 Oct 13. Food Chem Toxicol. 2023. PMID: 37839787
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
-
Mitochondrial Dynamics in Brain Cells During Normal and Pathological Aging.Int J Mol Sci. 2024 Nov 29;25(23):12855. doi: 10.3390/ijms252312855. Int J Mol Sci. 2024. PMID: 39684566 Free PMC article. Review.
-
Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes.Acta Med Okayama. 2016 Jun;70(3):151-8. doi: 10.18926/AMO/54413. Acta Med Okayama. 2016. PMID: 27339203 Review.
Cited by
-
Immunomodulatory Effects of Copper Bis-Glycinate In Vitro.Molecules. 2025 Mar 13;30(6):1282. doi: 10.3390/molecules30061282. Molecules. 2025. PMID: 40142058 Free PMC article.
-
The emerging role of cuproptosis in spinal cord injury.Front Immunol. 2025 Jun 16;16:1595852. doi: 10.3389/fimmu.2025.1595852. eCollection 2025. Front Immunol. 2025. PMID: 40589743 Free PMC article. Review.
-
Evolocumab attenuates myocardial ischemia/reperfusion injury by blocking PCSK9/LIAS-mediated cuproptosis of cardiomyocytes.Basic Res Cardiol. 2025 Apr;120(2):301-320. doi: 10.1007/s00395-025-01100-5. Epub 2025 Feb 11. Basic Res Cardiol. 2025. PMID: 39930254
-
Pathways to the Brain: Impact of Fine Particulate Matter Components on the Central Nervous System.Antioxidants (Basel). 2025 Jun 14;14(6):730. doi: 10.3390/antiox14060730. Antioxidants (Basel). 2025. PMID: 40563362 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous