

Talin1 dysfunction is genetically linked to systemic capillary leak syndrome
- PMID: 39704176
- PMCID: PMC11665552
- DOI: 10.1172/jci.insight.173664
Talin1 dysfunction is genetically linked to systemic capillary leak syndrome
Abstract
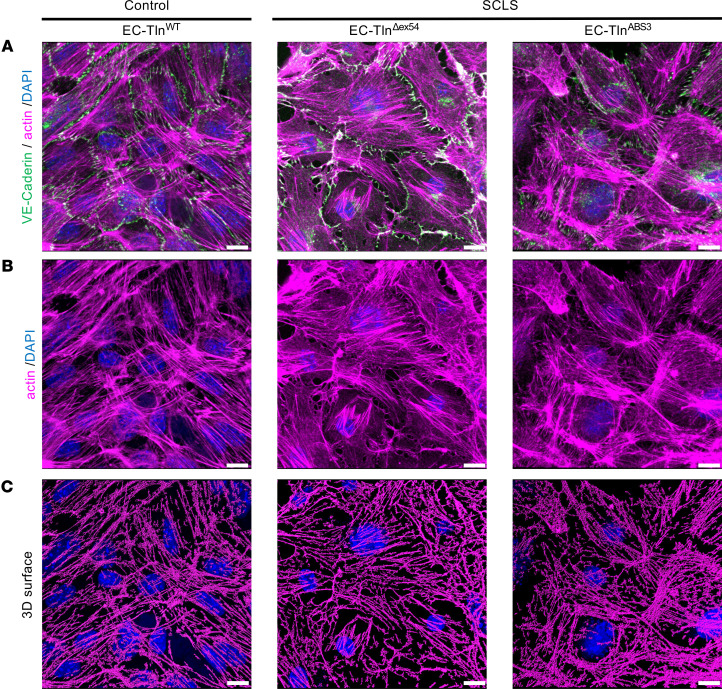
Systemic capillary leak syndrome (SCLS) is a rare life-threatening disorder due to profound vascular leak. The trigger and the cause of the disease are currently unknown and there is no specific treatment. Here, we identified a rare heterozygous splice-site variant in the TLN1 gene in a familial SCLS case, suggestive of autosomal dominant inheritance with incomplete penetrance. Talin1 has a key role in cell adhesion by activating and linking integrins to the actin cytoskeleton. This variant causes in-frame skipping of exon 54 and is predicted to affect talin's C-terminal actin-binding site (ABS3). Modeling the SCLS-TLN1 variant in TLN1-heterozygous endothelial cells (ECs) disturbed the endothelial barrier function. Similarly, mimicking the predicted actin-binding disruption in TLN1-heterozygous ECs resulted in disorganized endothelial adherens junctions. Mechanistically, we established that the SCLS-TLN1 variant, through the disruption of talin's ABS3, sequestrates talin's interacting partner, vinculin, at cell-extracellular matrix adhesions, leading to destabilization of the endothelial barrier. We propose that pathogenic variants in TLN1 underlie SCLS, providing insight into the molecular mechanism of the disease that can be explored for future therapeutic interventions.
Keywords: Cell biology; Cell migration/adhesion; Endothelial cells; Integrins; Vascular biology.
Figures







References
-
- Clarkson B, et al. Cyclical edema and shock due to increased capillary permeability. Trans Assoc Am Physicians. 1960;73(2):272–282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
