

CBL0137 and NKG2A blockade: a novel immuno-oncology combination therapy for Myc-overexpressing triple-negative breast cancers
- PMID: 39706891
- PMCID: PMC11932921
- DOI: 10.1038/s41388-024-03259-y
CBL0137 and NKG2A blockade: a novel immuno-oncology combination therapy for Myc-overexpressing triple-negative breast cancers
Erratum in
-
Correction: CBL0137 and NKG2A blockade: a novel immuno-oncology combination therapy for Myc-overexpressing triple-negative breast cancers.Oncogene. 2025 Apr;44(13):927. doi: 10.1038/s41388-025-03319-x. Oncogene. 2025. PMID: 40033028 Free PMC article. No abstract available.
Abstract
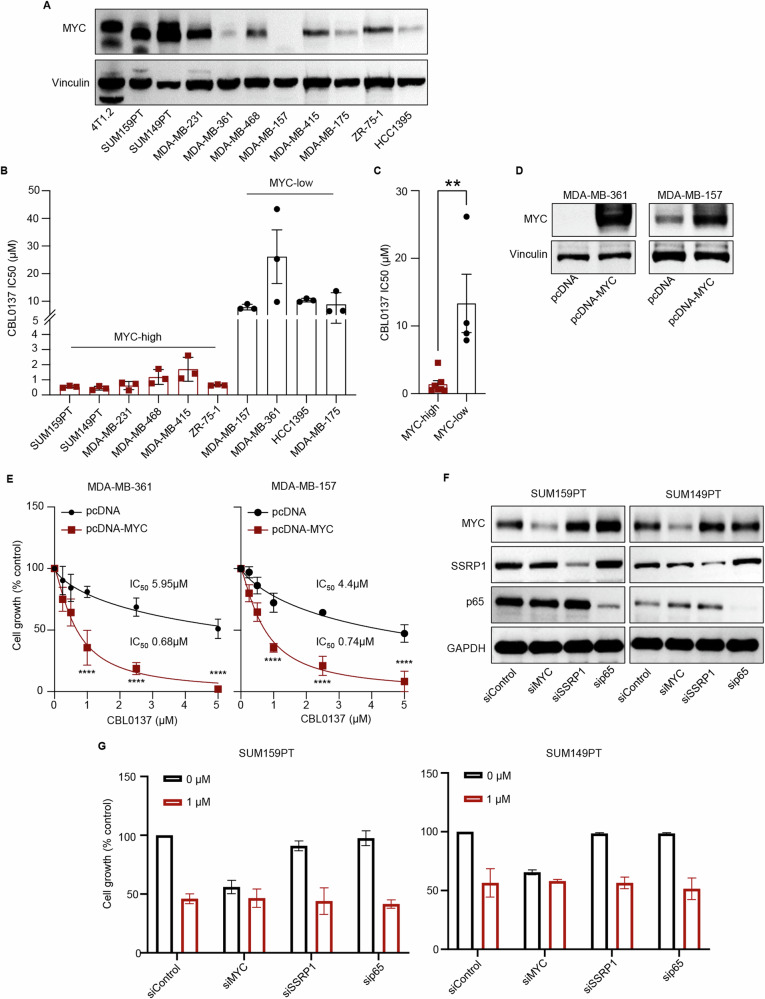
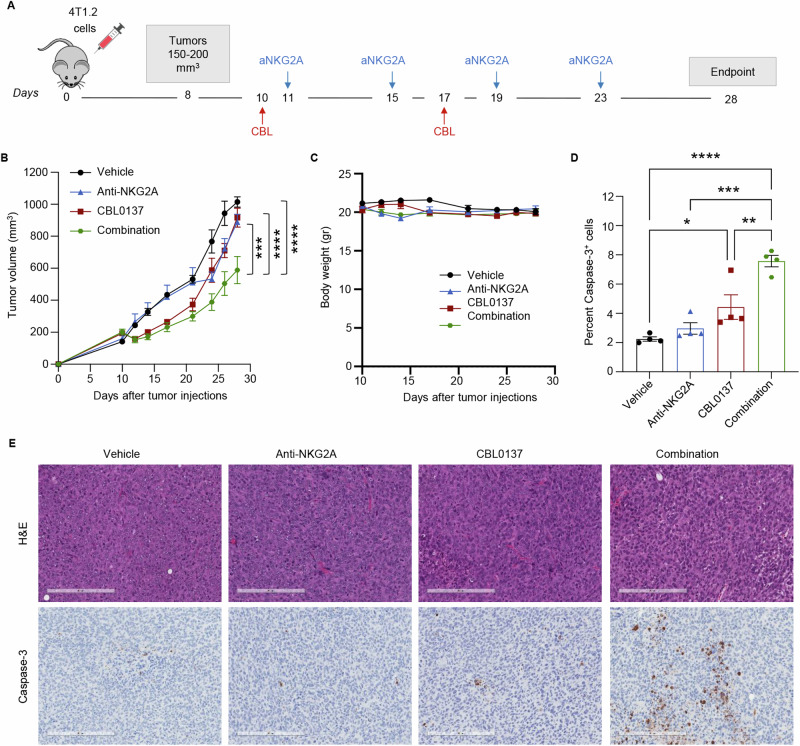
The MYC proto-oncogene is upregulated in >60% of triple-negative breast cancers (TNBCs), it can directly promote tumor cell proliferation, and its overexpression negatively regulates anti-tumor immune responses. For all these reasons, MYC has long been considered as a compelling therapeutic target. However, pharmacological inhibition of MYC function has proven difficult due to a lack of a drug-binding pocket. Here, we demonstrate that the potent abrogation of MYC gene transcription by CBL0137 induces immunogenic cell death and reduces proliferation in MYC-high but not in MYC-low TNBC in vitro. CBL0137 also significantly inhibited the in vivo growth of primary tumors in a human MYC-high TNBC xenograft model (MDA-MB-231). Moreover, CBL0137 inhibited the tumor growth of highly aggressive mouse 4T1.2 syngeneic TNBC model in immunocompetent mice by inhibiting the MYC pathway and inducing Type I interferon responses. Immune profiling of CBL0137-treated mice revealed significantly enhanced tumor-specific immune responses and increased proportions of tumor infiltrating effector CD8+ T cells, CD4+ T cells, and NK cells. CBL0137-induced immune activation also resulted in increased exhaustion of immune effector cells. In particular, NKG2A up-regulation on activated effector cells and of its ligand Qa-1b on tumors in vivo was identified as a possible immune evasive mechanism. Indeed, NKG2A blockade synergized with CBL0137 significantly inhibiting the in vivo growth of 4T1.2 tumors. Collectively, our findings provide the rationale supporting the exploitation of CBL0137-induced anti-tumor immunity in combination with NKG2A blockade to improve the treatment of TNBC expressing high levels of MYC.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics Declarations: All experimental methods were performed in accordance with the guidelines of the QIMR Berghofer Medical Research Institute Animal Ethics Committee Approved ethics: A2108-613, the Standard Operating Procedure (SOPs) approved by the University of Queensland Animal Ethic Committee (UQ Health Sciences Ethics Committee (HS) Approved Ethics: UQDI/252/16), and the guidelines of the Peter MacCallum Cancer Center Animal Ethics Committee (Approved ethics: XXX).
Figures





References
-
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. - PubMed
-
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–4. - PubMed
-
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
