

Targeted long-read sequencing to quantify methylation of the C9orf72 repeat expansion
- PMID: 39709476
- PMCID: PMC11663317
- DOI: 10.1186/s13024-024-00790-0
Targeted long-read sequencing to quantify methylation of the C9orf72 repeat expansion
Abstract
Background: The gene C9orf72 harbors a non-coding hexanucleotide repeat expansion known to cause amyotrophic lateral sclerosis and frontotemporal dementia. While previous studies have estimated the length of this repeat expansion in multiple tissues, technological limitations have impeded researchers from exploring additional features, such as methylation levels.
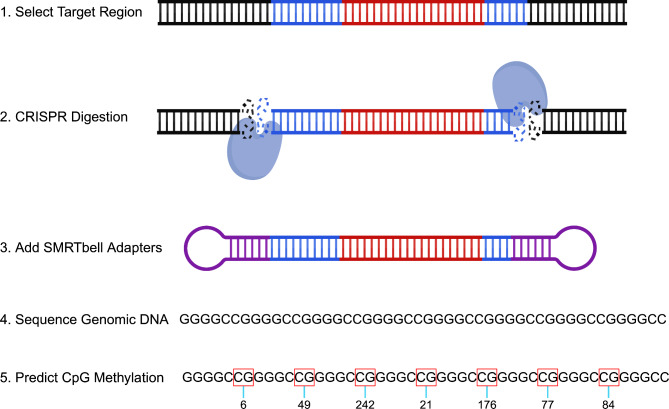
Methods: We aimed to characterize C9orf72 repeat expansions using a targeted, amplification-free long-read sequencing method. Our primary goal was to determine the presence and subsequent quantification of observed methylation in the C9orf72 repeat expansion. In addition, we measured the repeat length and purity of the expansion. To do this, we sequenced DNA extracted from blood for 27 individuals with an expanded C9orf72 repeat.
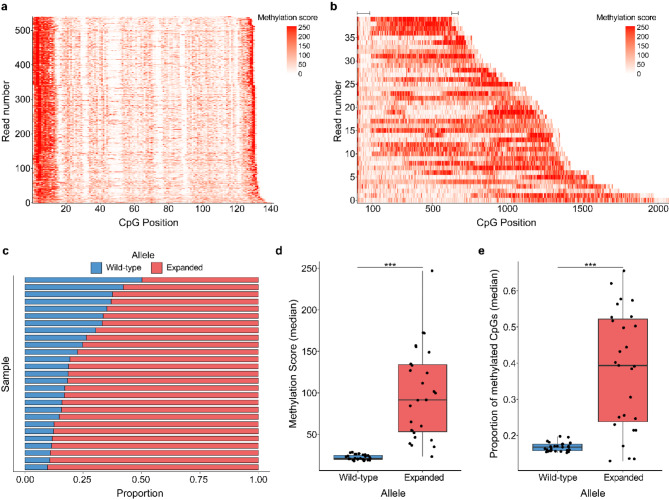
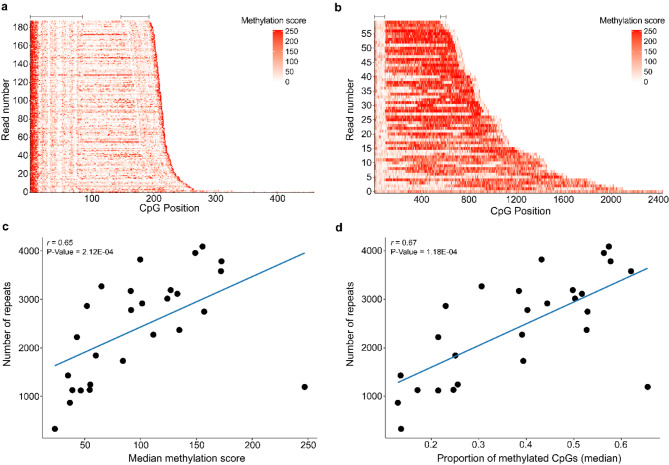
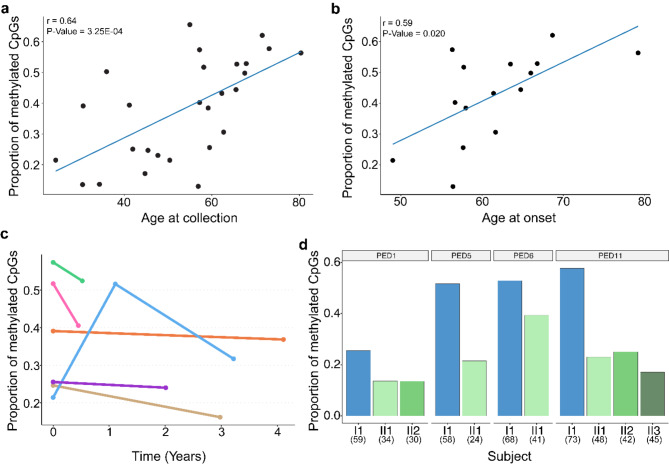
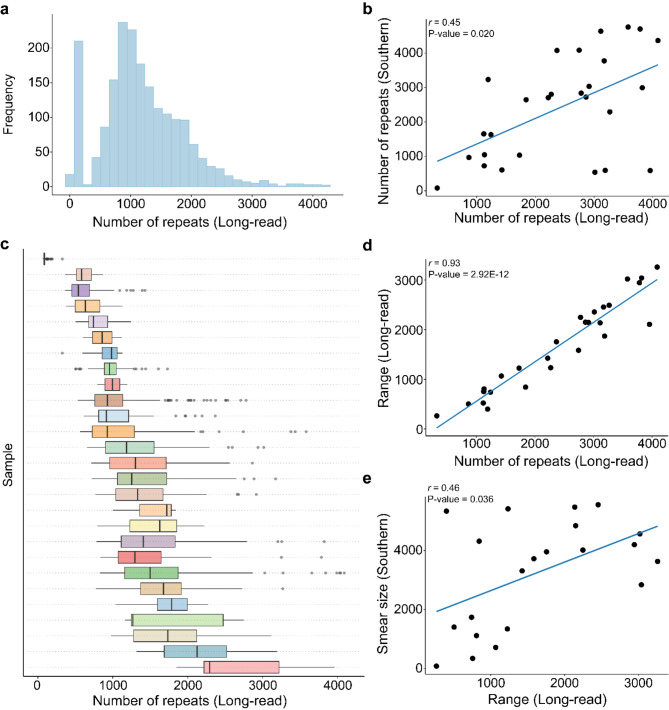
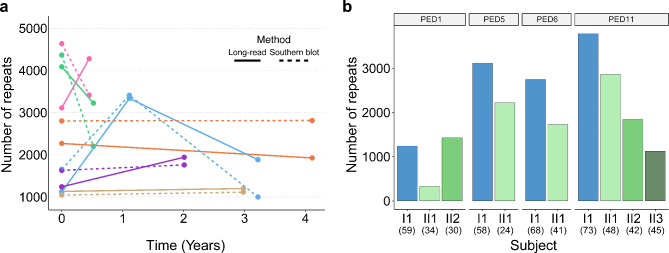
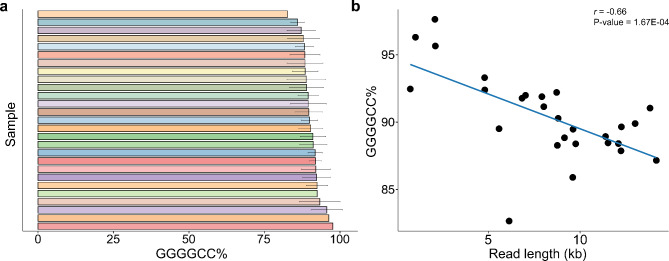
Results: For these individuals, we obtained a total of 7,765 on-target reads, including 1,612 fully covering the expanded allele. Our in-depth analysis revealed that the expansion itself is methylated, with great variability in total methylation levels observed, as represented by the proportion of methylated CpGs (13 to 66%). Interestingly, we demonstrated that the expanded allele is more highly methylated than the wild-type allele (P-Value = 2.76E-05) and that increased methylation levels are observed in longer repeat expansions (P-Value = 1.18E-04). Furthermore, methylation levels correlate with age at collection (P-Value = 3.25E-04) as well as age at disease onset (P-Value = 0.020). Additionally, we detected repeat lengths up to 4,088 repeats (~ 25 kb) and found that the expansion contains few interruptions in the blood.
Conclusions: Taken together, our study demonstrates robust ability to quantify methylation of the expanded C9orf72 repeat, capturing differences between individuals harboring this expansion and revealing clinical associations.
Keywords: Amyotrophic lateral sclerosis; C9orf72; Frontotemporal dementia; Long-read sequencing; Methylation; Repeat expansions.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All subjects agreed to be in the study, and biological specimens were obtained after informed consent with approval from the Mayo Clinic Institutional Review Board (IRB). Consent for publication: Not applicable. Competing interests: MDJ and RR hold a patent on methods to screen for the C9orf72 hexanucleotide repeat expansion.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
