

This is a preprint.
Design of allosteric modulators that change GPCR G protein subtype selectivity
- PMID: 39711540
- PMCID: PMC11661308
- DOI: 10.21203/rs.3.rs-5538058/v1
Design of allosteric modulators that change GPCR G protein subtype selectivity
Update in
-
Designing allosteric modulators to change GPCR G protein subtype selectivity.Nature. 2025 Dec;648(8092):229-238. doi: 10.1038/s41586-025-09643-2. Epub 2025 Oct 22. Nature. 2025. PMID: 41125894 Free PMC article.
Abstract
G protein-coupled receptors (GPCRs), the largest family of drug targets, can signal through 16 subtypes of Gα proteins. Biased compounds that selectively activate therapy-relevant pathways promise to be safer, more effective medications. The determinants of bias are poorly understood, however, and rationally-designed, G protein-subtype-selective compounds are lacking. Here, using the prototypical class A GPCR neurotensin receptor 1 (NTSR1), we find that small molecules binding the intracellular GPCR-transducer interface change G protein coupling by subtype-specific and predictable mechanisms, enabling rational drug design. We demonstrate that the compound SBI-553 switches NTSR1 G protein preference by acting both as a molecular bumper and a molecular glue. Structurally, SBI-553 occludes G protein binding determinants on NTSR1, promoting association with select G protein subtypes for which an alternative, shallow-binding conformation is energetically favorable. Minor modifications to the SBI-553 scaffold produce allosteric modulators with distinct G protein subtype selectivity profiles. Selectivity profiles are probe-independent, conserved across species, and translate to differences in in vivo activity. These studies demonstrate that G protein selectivity can be tailored with small changes to a single chemical scaffold targeting the receptor-transducer interface and, as this pocket is broadly conserved, present a strategy for pathway-selective drug discovery applicable to the diverse GPCR superfamily.
Keywords: G protein selectivity; G protein-coupled receptor; allosteric modulation; biased signaling; neurotensin receptor 1; β-arrestin.
Figures


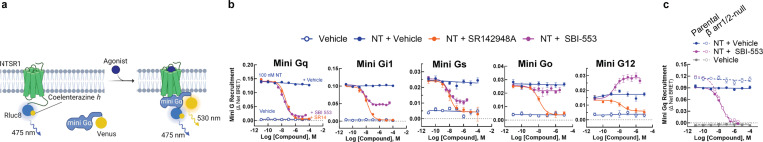

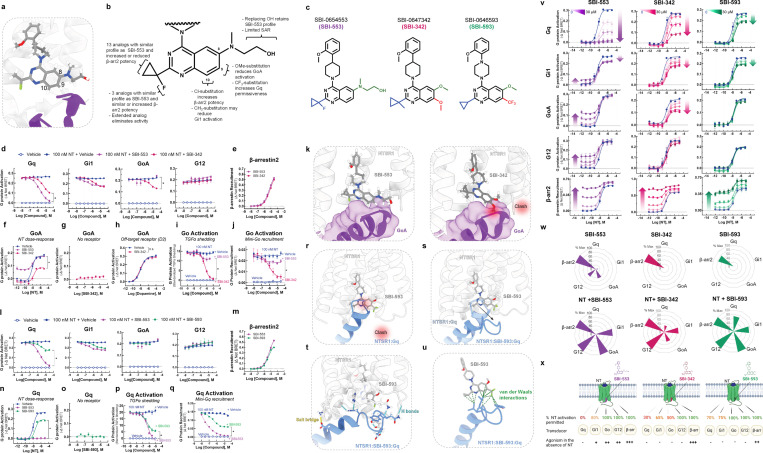
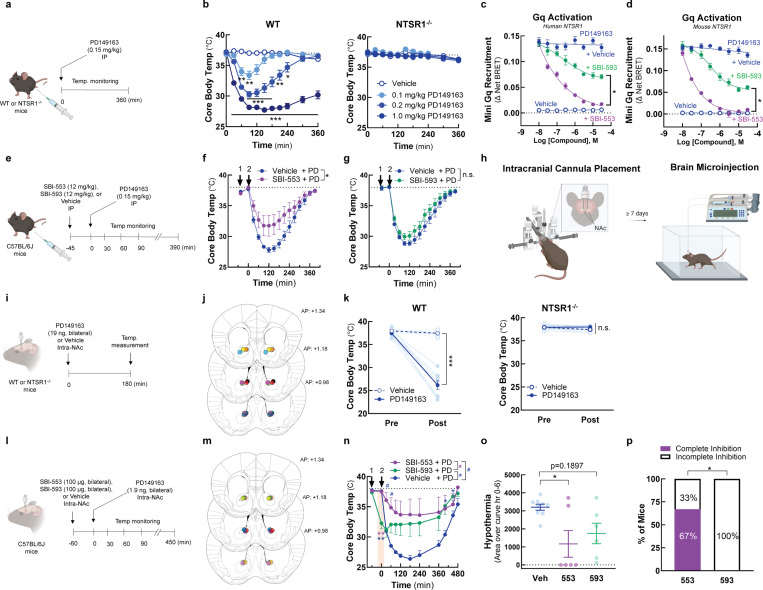
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
