

Pulmonary arterial hypertension with left to right shunts: When to treat and/or close?
- PMID: 39711769
- PMCID: PMC11657717
- DOI: 10.1016/j.ijcchd.2024.100526
Pulmonary arterial hypertension with left to right shunts: When to treat and/or close?
Abstract
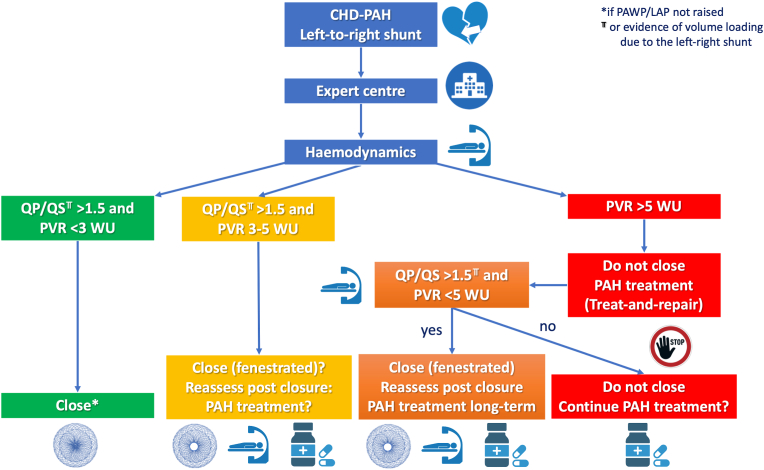
Pulmonary arterial hypertension (PAH) is defined as increase in mean pulmonary arterial pressure and pulmonary vascular resistance (PVR). It can be associated with congenital heart disease (CHD) with the following subtypes: 1) uncorrected left-to-right (L-R) intracardiac shunt leading to overload of the pulmonary circulation and a progressive increase of PVR; 2) Eisenmenger syndrome, appearing when a large post-tricuspid shunt is left uncorrected and pulmonary vascular disease (PVD) is severe, so the shunt becomes bidirectional or right-to-left, causing cyanosis; 3) PAH after shunt closure, when PVR arises after a defect correction; and 4) PAH associated with small or coincidental defects. While the treatment of patients with Eisenmenger syndrome is well established, the treatment of patients with PAH in whom there is a L-R shunt (with no cyanosis) remains unclear and requires expertise. In such patients, correction of the defect may be contemplated if there is mild PVD and a significant L-R shunt. Others may benefit from a "treat and repair" strategy, which involves the use of PAH therapy to achieve a drop in PVR, with the aim of achieving operability criteria. Cardiac catheterization is at the center of the evaluation and follow-up of these patients, collecting "baseline" data and providing the opportunity to challenge the pulmonary circulation, manipulate the loading status, or temporarily occlude the defect. This article provides a detailed overview of the pathophysiology and treatment options for patients with PAH associated with a L-R congenital shunt, including current approaches to operability and the use of PAH therapies.
Keywords: Congenital heart disease; Hemodynamics assessment; Intracardiac shunts; Pulmonary hypertension.
© 2024 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper, other than two of them (MA and KD) serving in the IJCCHD Editorila Board.
Figures
References
-
- Humbert M., Kovacs G., Hoeper M.M., et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43:3618–3731. - PubMed
-
- Beghetti M., Galie` N. Eisenmenger syndrome a clinical perspective in a new therapeutic era of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;53:733–740. - PubMed
-
- Collins-Nakai R.L., Rabinovitch M. Pulmonary vascular obstructive disease. Cardiol Clin. 1993;11:675–687. - PubMed
-
- Duffels M.G., Engelfriet P.M., Berger R.M., et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol. 2007;120:198–204. - PubMed
-
- Greenwood R.D., Nadas A.S. The clinical course of cardiac disease in Down's syndrome. Pediatrics. 1976;58:893–897. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
