

Remodeling of the chromatin landscape in peripheral blood cells in patients with severe Delta COVID-19
- PMID: 39712003
- PMCID: PMC11662282
- DOI: 10.3389/fimmu.2024.1415317
Remodeling of the chromatin landscape in peripheral blood cells in patients with severe Delta COVID-19
Abstract
COVID-19 is characterized by systemic pro-inflammatory shifts with the development of serious alterations in the functioning of the immune system. Investigations of the gene expression changes accompanying the infection state provide insight into the molecular and cellular processes depending on the sickness severity and virus variants. Severe Delta COVID-19 has been characterized by the appearance of a monocyte subset enriched for proinflammatory gene expression signatures and a shift in ligand-receptor interactions. We profiled the chromatin accessibility landscape of 140,000 nuclei in PBMC samples from healthy individuals or individuals with COVID-19. We investigated cis-regulatory elements and identified the core transcription factors governing gene expression in immune cells during COVID-19 infection. In severe cases, we discovered that regulome and chromatin co-accessibility modules were significantly altered across many cell types. Moreover, cases with the Delta variant were accompanied by a specific monocyte subtype discovered using scATAC-seq data. Our analysis showed that immune cells of individuals with severe Delta COVID-19 underwent significant remodeling of the chromatin accessibility landscape and development of the proinflammatory expression pattern. Using a gene regulatory network modeling approach, we investigated the core transcription factors governing the cell state and identified the most pronounced chromatin changes in CD14+ monocytes from individuals with severe Delta COVID-19. Together, our results provide novel insights into cis-regulatory module organization and its impact on gene activity in immune cells during SARS-CoV-2 infection.
Keywords: COVID-19; PBMC (peripheral blood mononuclear cells); scATAC-seq; single cell; transcriptional regulatory network.
Copyright © 2024 Akimov, Tychinin, Antonova, Shaymardanov, Voronina, Deinichenko, Fateev, Yudin, Yudin, Mukhin, Romanova, Nekrasova, Zhdanova, Tsypkina, Vladimirov, Makhotenko, Keskinov, Kraevoy, Snigir, Svetlichnyy and Skvortsova.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
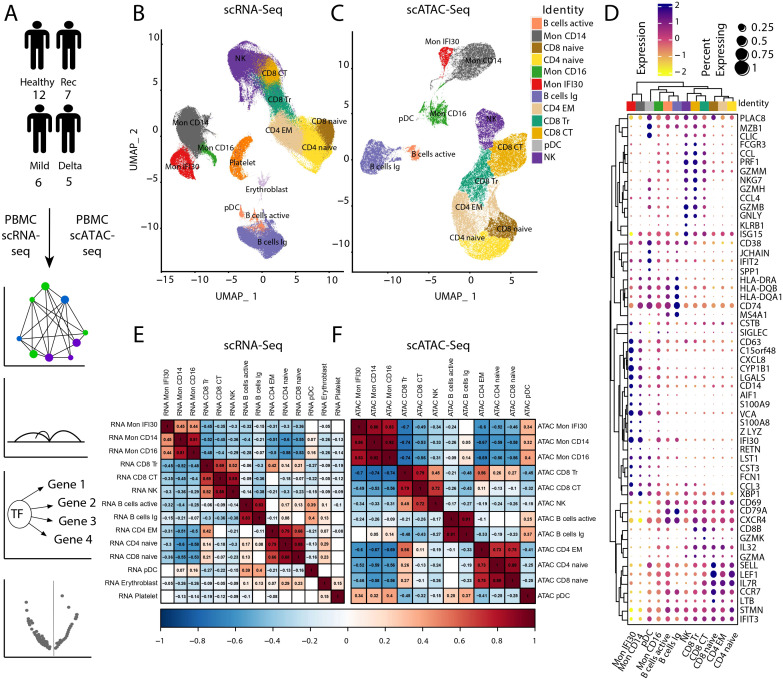
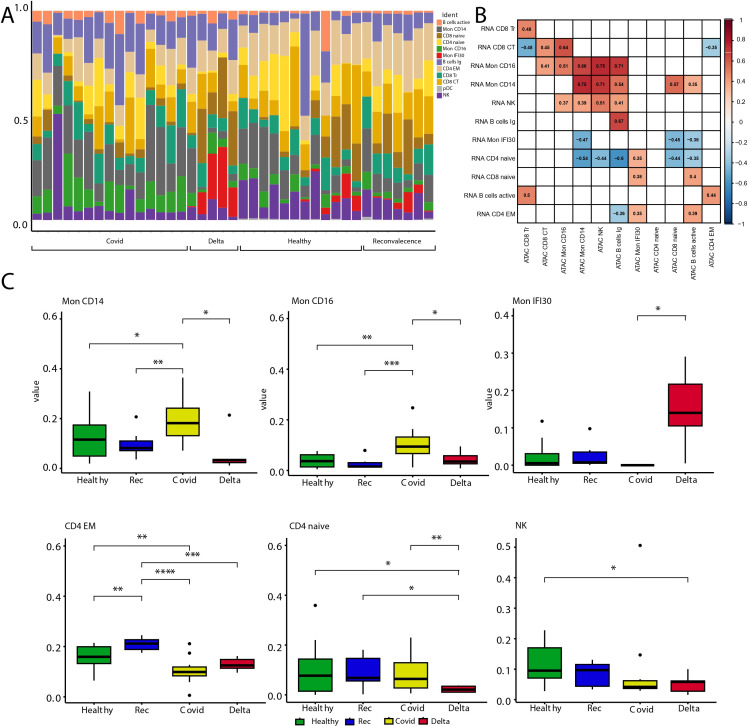
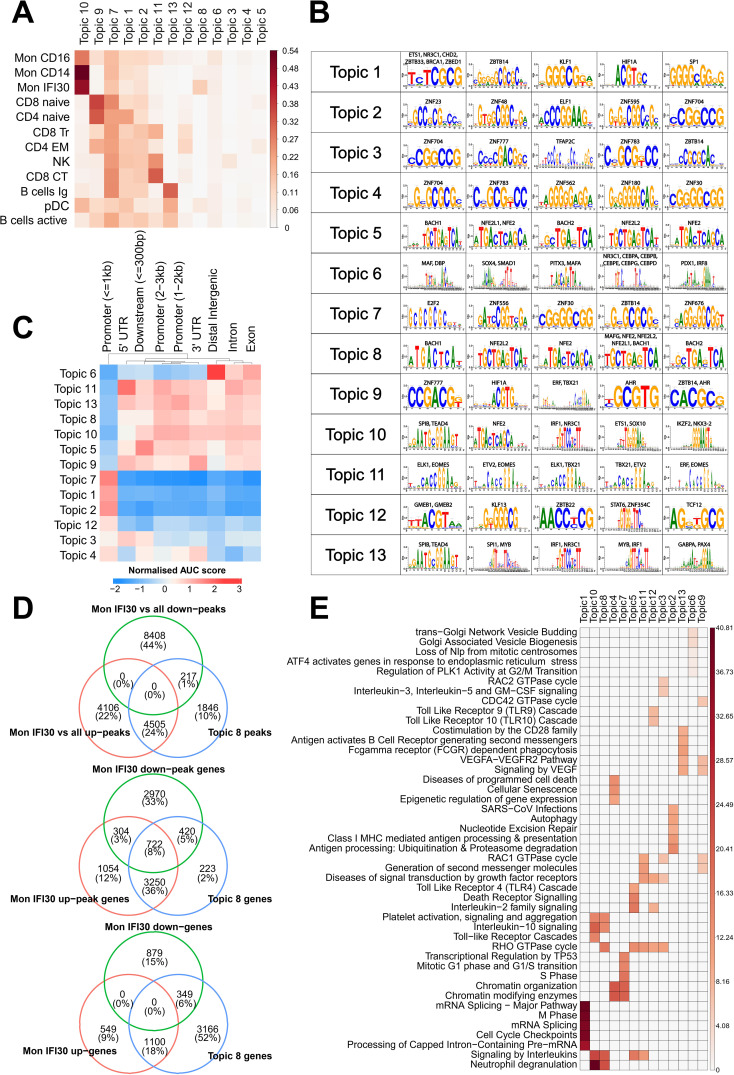
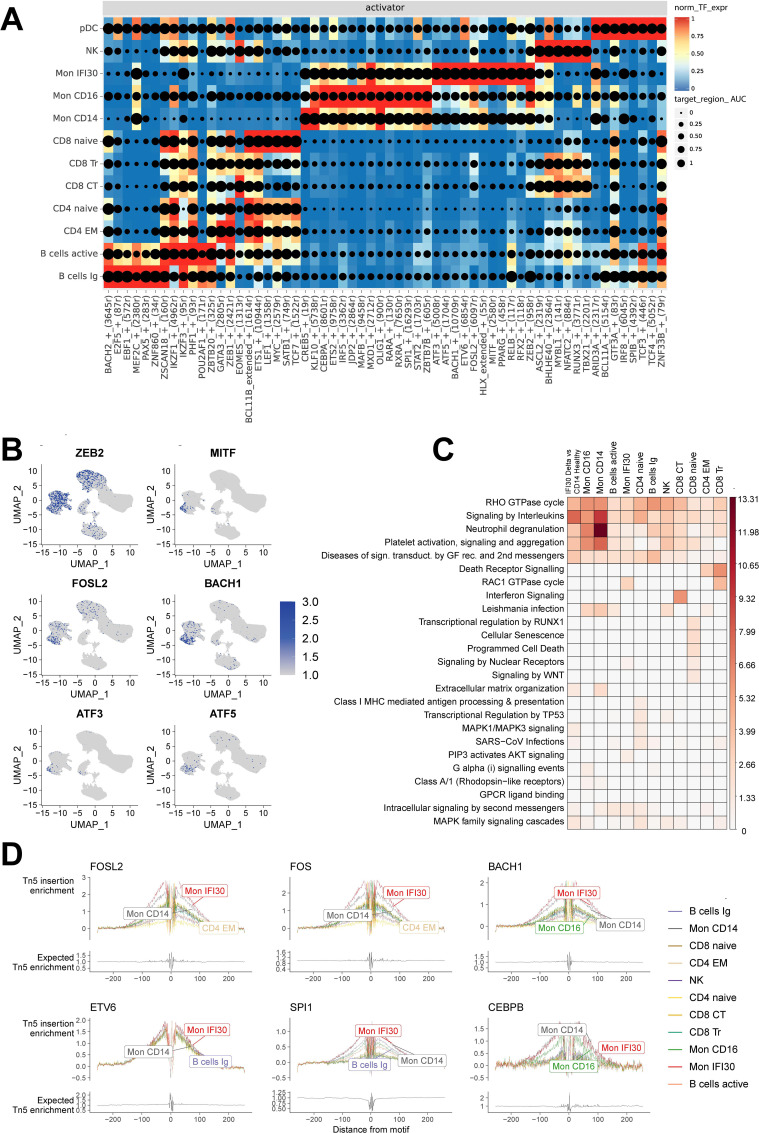
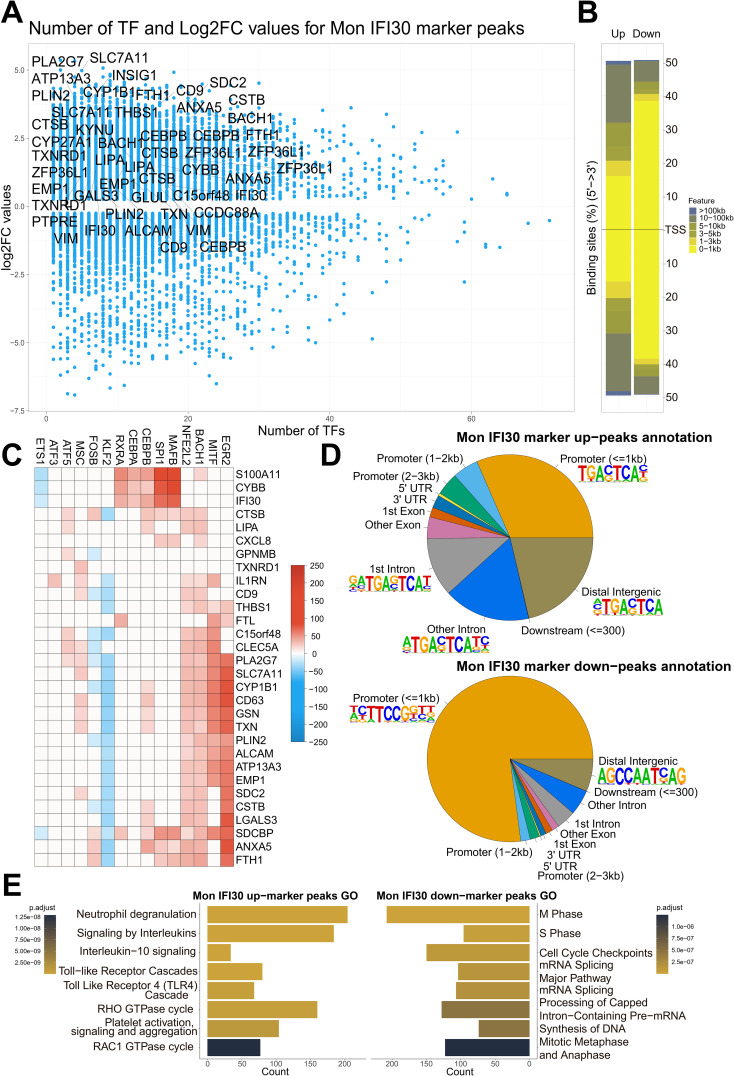
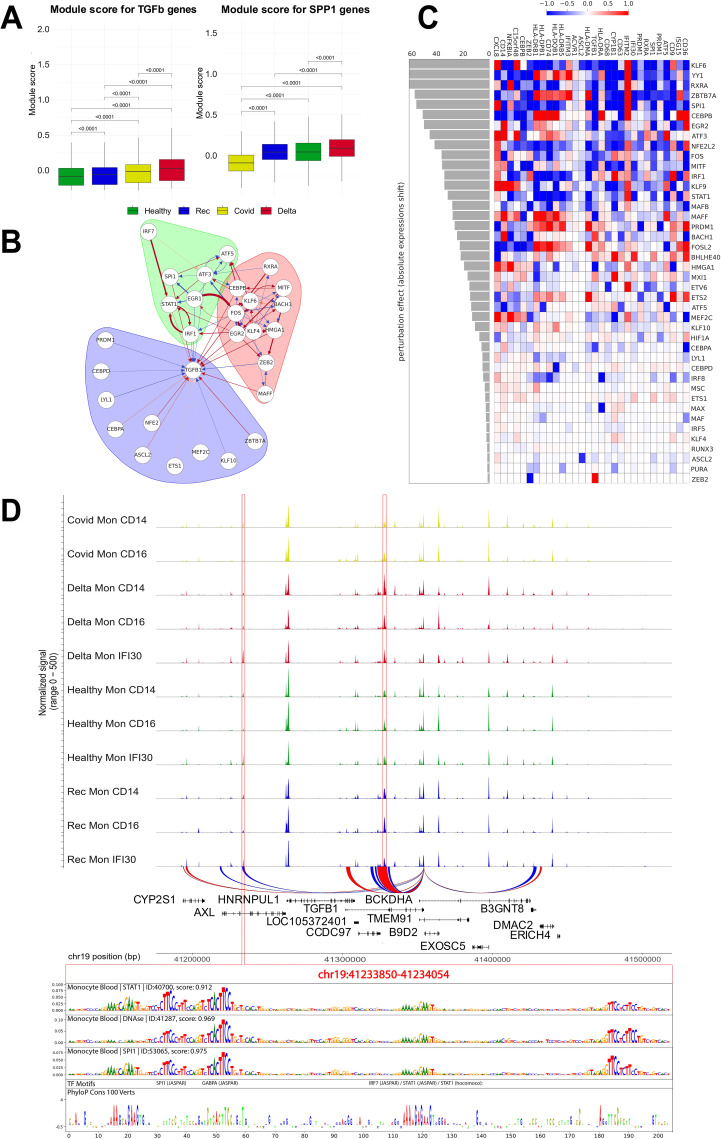
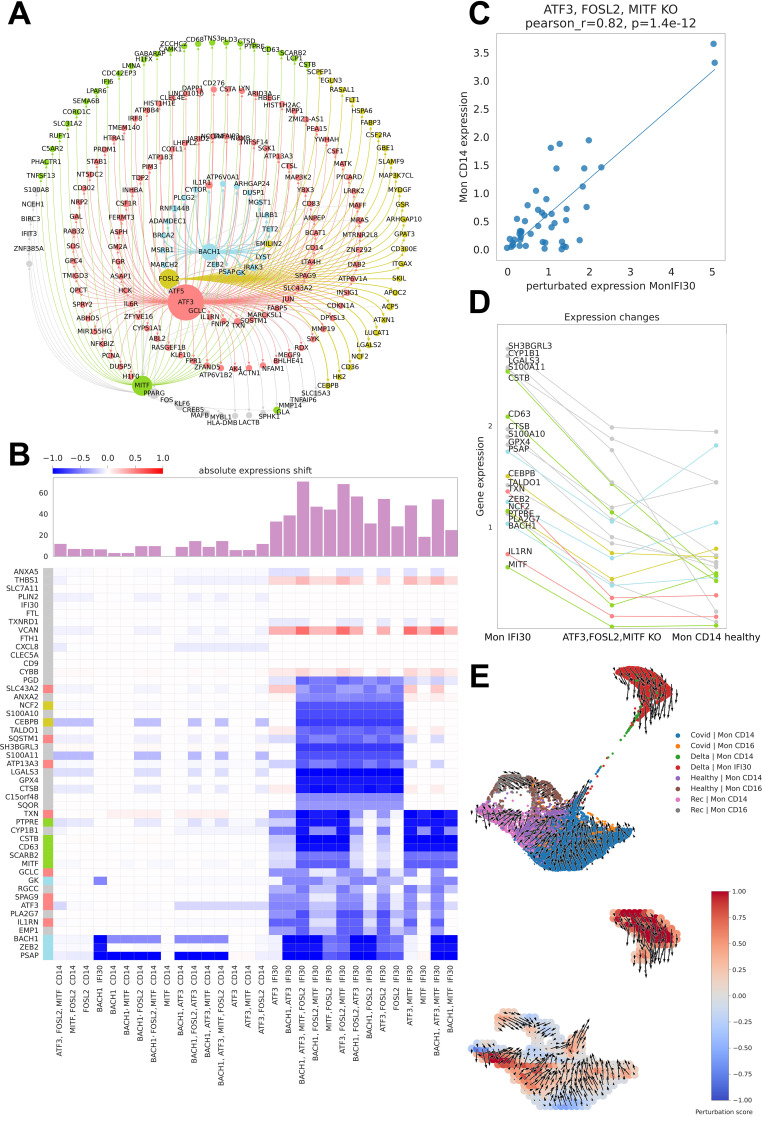
References
-
- Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal Complex Syst. (2006) 1695:1–9. doi: 10.5281/zenodo.7682609 - DOI
-
- Shrikumar A, Greenside P, Kundaje A. Learning important features through propagating activation differences. arXiv: 1704.02685. (2017) 3145–53. doi: 10.48550/arXiv.1704.02685 - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous