

Integrative multi-omics analysis reveals a novel subtype of hepatocellular carcinoma with biological and clinical relevance
- PMID: 39712016
- PMCID: PMC11659151
- DOI: 10.3389/fimmu.2024.1517312
Integrative multi-omics analysis reveals a novel subtype of hepatocellular carcinoma with biological and clinical relevance
Abstract
Background: Hepatocellular carcinoma (HCC) is a highly heterogeneous tumor, and the development of accurate predictive models for prognosis and drug sensitivity remains challenging.
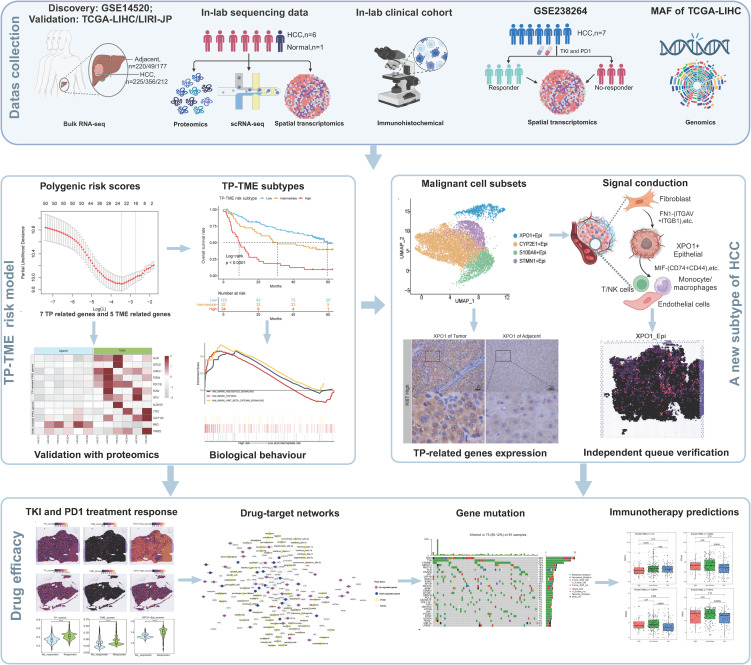
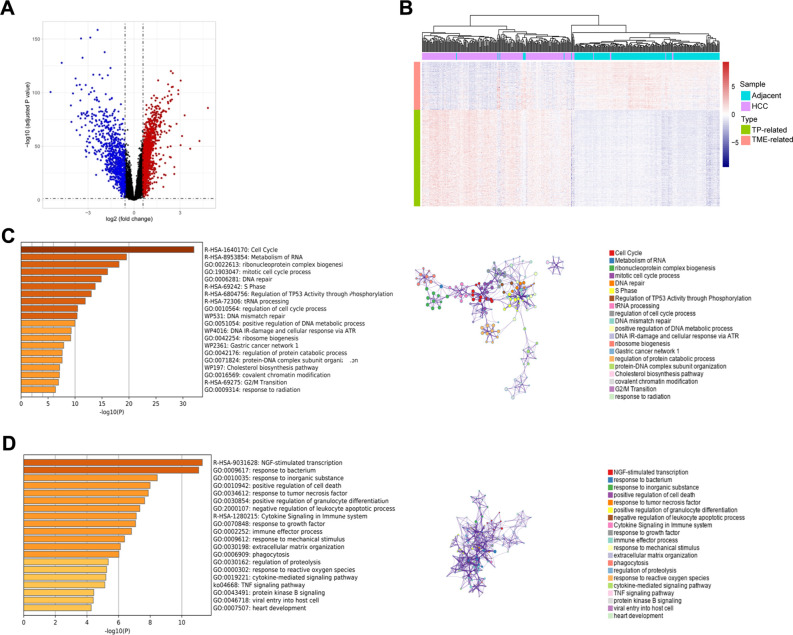
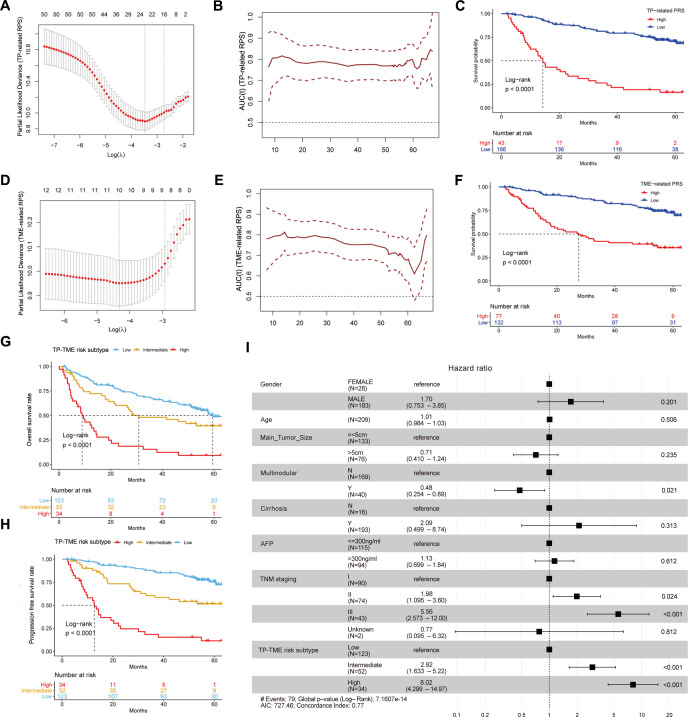
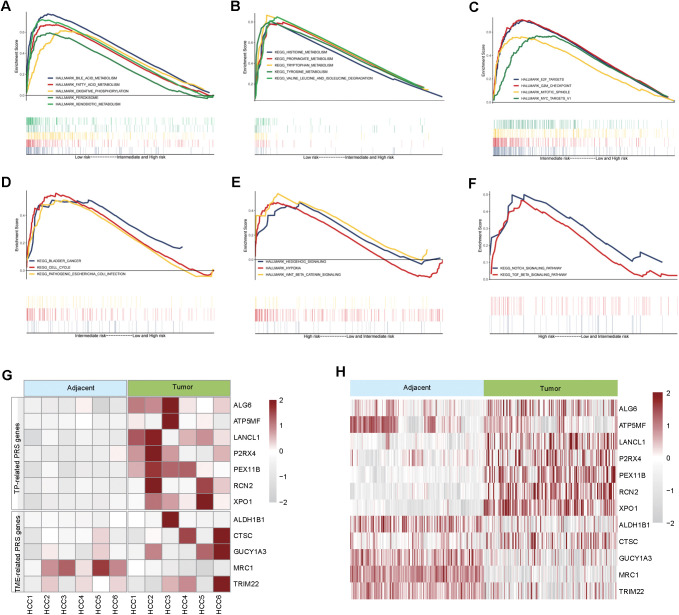
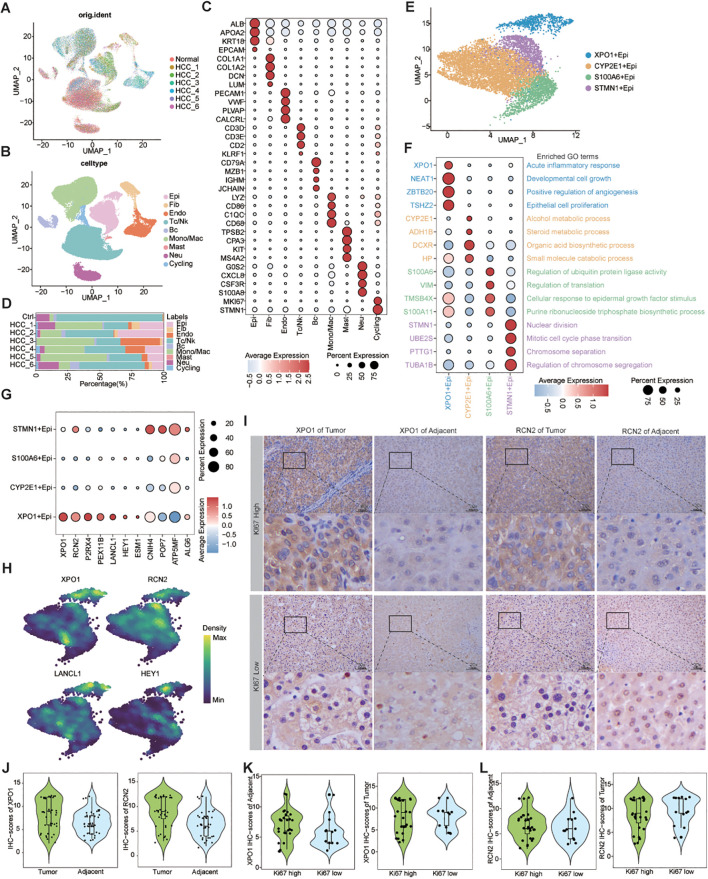
Methods: We integrated laboratory data and public cohorts to conduct a multi-omics analysis of HCC, which included bulk RNA sequencing, proteomic analysis, single-cell RNA sequencing (scRNA-seq), spatial transcriptomics sequencing (ST-seq), and genome sequencing. We constructed a tumor purity (TP) and tumor microenvironment (TME) prognostic risk model. Proteomic analysis validated the TP-TME-related signatures. Joint analysis of scRNA-seq and ST-seq revealed characteristic clusters associated with TP high-risk subtypes, and immunohistochemistry confirmed the expression of key genes. We conducted functional enrichment analysis, transcription factor activity inference, cell-cell interaction, drug efficacy analysis, and mutation information analysis to identify a novel subtype of HCC.
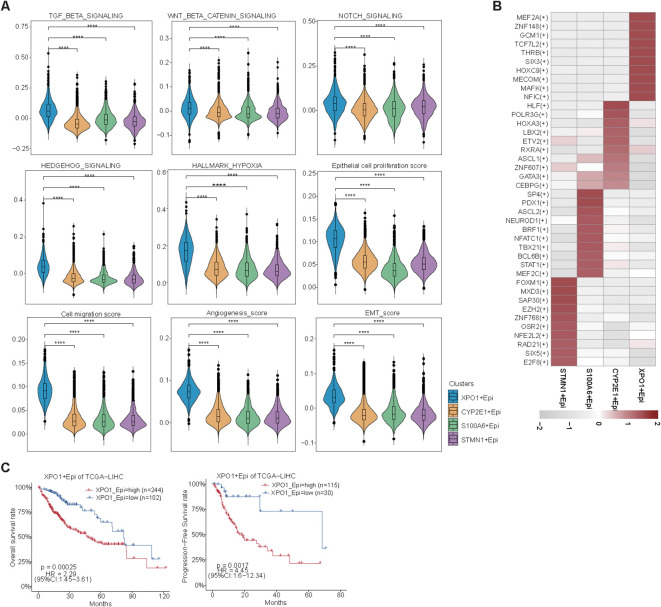
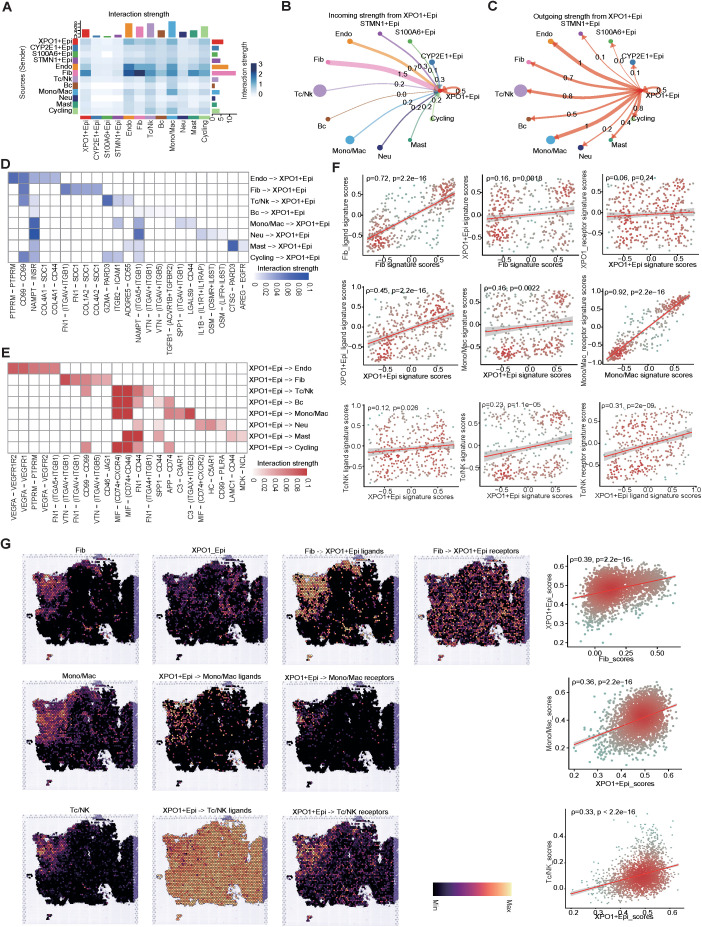
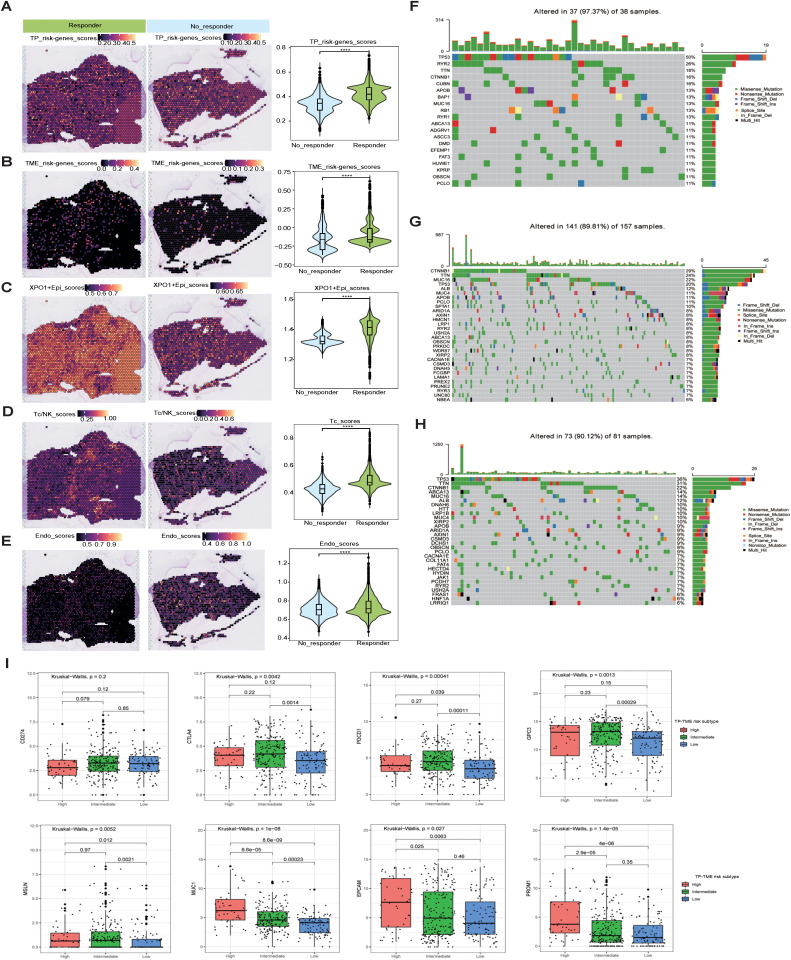
Results: Our analyses constructed a robust HCC prognostic risk prediction model. The patients with TP-TME high-risk subtypes predominantly exhibit hypoxia and activation of the Wnt/beta-catenin, Notch, and TGF-beta signaling pathways. Furthermore, we identified a novel subtype, XPO1+Epithelial. This subtype expresses signatures of the TP risk subtype and aligns with the biological behavior of high-risk patients. Additional analyses revealed that XPO1+Epithelial is influenced primarily by fibroblasts via ligand-receptor interactions, such as FN1-(ITGAV+ITGB1), and constitute a significant component of the TP-TME subtype. Moreover, XPO1+Epithelial interact with monocytes/macrophages, T/NK cells, and endothelial cells through ligand-receptor pairs, including MIF-(CD74+CXCR4), MIF-(CD74+CD44), and VEGFA-VEGFR1R2, respectively, thereby promoting the recruitment of immune-suppressive cells and angiogenesis. The ST-seq cohort treated with Tyrosine Kinase Inhibitors (TKIs) and Programmed Cell Death Protein 1 (PD-1) presented elevated levels of TP and TME risk subtype signature genes, as well as XPO1+Epithelial, T-cell, and endothelial cell infiltration in the treatment response group. Drug sensitivity analyses indicated that TP-TME high-risk subtypes, including sorafenib and pembrolizumab, were associated with sensitivity to multiple drugs. Further exploratory analyses revealed that CTLA4, PDCD1, and the cancer antigens MSLN, MUC1, EPCAM, and PROM1 presented significantly increase expression levels in the high-risk subtype group.
Conclusions: This study constructed a robust prognostic model for HCC and identified novel subgroups at the single-cell level, potentially assisting in the assessment of prognostic risk for HCC patients and facilitating personalized drug therapy.
Keywords: hepatocellular carcinoma; immunotherapy; precision medicine; single-cell RNA sequencing; spatial transcriptomics; tumor microenvironment; tumor purity.
Copyright © 2024 Li, Lin, Gao, Zeng, Cen, Su, Su, Zeng, Huang, Zeng, Huang, Tang, Li, Luo, Huang, Liang and Ye.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous