

This is a preprint.
Pol II degradation activates cell death independently from the loss of transcription
- PMID: 39713309
- PMCID: PMC11661175
- DOI: 10.1101/2024.12.09.627542
Pol II degradation activates cell death independently from the loss of transcription
Update in
-
RNA Pol II inhibition activates cell death independently from the loss of transcription.Cell. 2025 Oct 30;188(22):6301-6316.e29. doi: 10.1016/j.cell.2025.07.034. Epub 2025 Aug 15. Cell. 2025. PMID: 40818455 Free PMC article.
Abstract
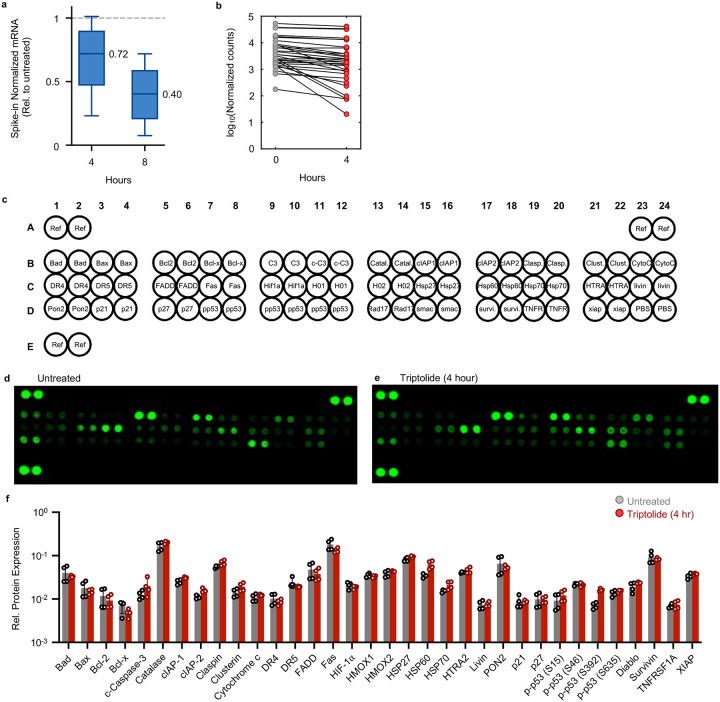
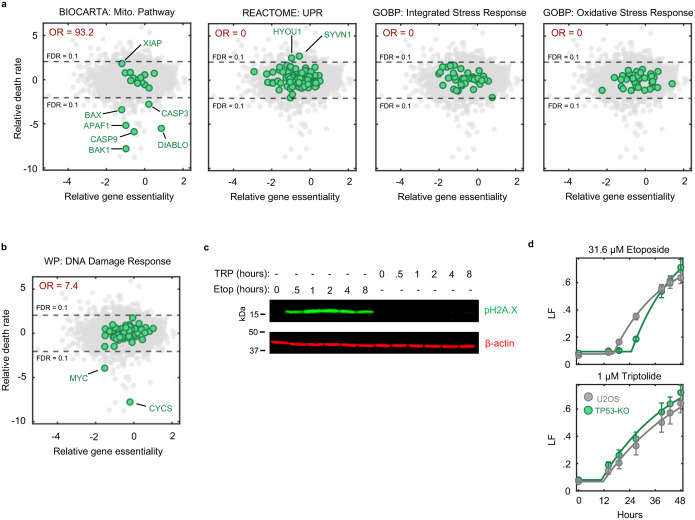
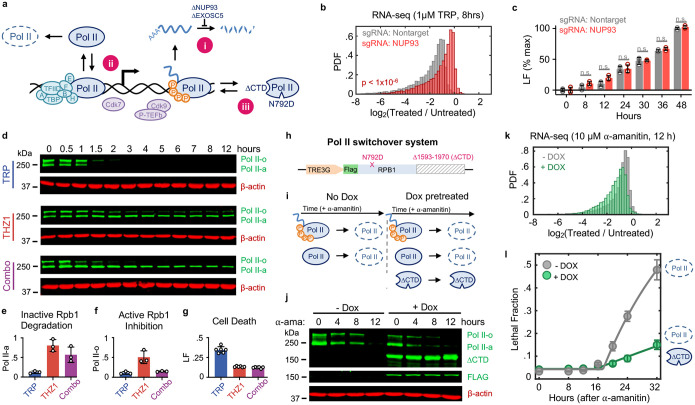
Pol II-mediated transcription is essential for eukaryotic life. While loss of transcription is thought to be universally lethal, the associated mechanisms promoting cell death are not yet known. Here, we show that death following loss of Pol II is not caused by dysregulated gene expression. Instead, death occurs in response to the loss of Pol II protein itself, specifically loss of the enzymatic subunit, Rbp1. Loss of Pol II exclusively activates apoptosis, and expression of a transcriptionally inactive version of Rpb1 rescues cell viability. Using functional genomics, we identify a previously uncharacterized mechanism that regulates lethality following loss of Pol II, which we call the Pol II Degradation-dependent Apoptotic Response (PDAR). Using the genetic dependencies of PDAR, we identify clinically used drugs that owe their efficacy to a PDAR-dependent mechanism. Our findings unveil a novel apoptotic signaling response that contributes to the efficacy of a wide array of anti-cancer therapies.
Conflict of interest statement
Competing interests The authors declare that they have no competing interests.
Figures


























References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous