

This is a preprint.
Alignment-Free Viral Sequence Classification at Scale
- PMID: 39713356
- PMCID: PMC11661207
- DOI: 10.1101/2024.12.10.627186
Alignment-Free Viral Sequence Classification at Scale
Update in
-
Alignment-free viral sequence classification at scale.BMC Genomics. 2025 Apr 18;26(1):389. doi: 10.1186/s12864-025-11554-5. BMC Genomics. 2025. PMID: 40251515 Free PMC article.
Abstract
Background: The rapid increase in nucleotide sequence data generated by next-generation sequencing (NGS) technologies demands efficient computational tools for sequence comparison. Alignment-based methods, such as BLAST, are increasingly overwhelmed by the scale of contemporary datasets due to their high computational demands for classification. This study evaluates alignment-free (AF) methods as scalable and rapid alternatives for viral sequence classification, focusing on identifying techniques that maintain high accuracy and efficiency when applied to extremely large datasets.
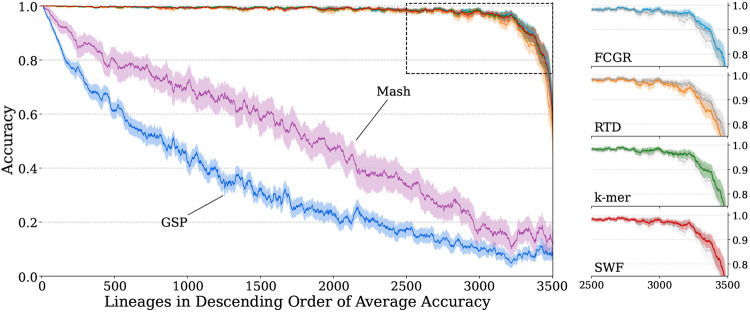
Results: We employed six established AF techniques to extract feature vectors from viral genomes, which were subsequently used to train Random Forest classifiers. Our primary dataset comprises 297,186 SARS-CoV-2 nucleotide sequences, categorized into 3502 distinct lineages. Furthermore, we validated our models using dengue and HIV sequences to demonstrate robustness across different viral datasets. Our AF classifiers achieved 97.8% accuracy on the SARS-CoV-2 test set, and 99.8% and 89.1% accuracy on dengue and HIV test sets, respectively.
Conclusion: Despite the high-class dimensionality, we show that word-based AF methods effectively represent viral sequences. Our study highlights the practical advantages of AF techniques, including significantly faster processing compared to alignment-based methods and the ability to classify sequences using modest computational resources.
Keywords: alignment-free; biological sequences; feature extraction; machine learning; virus classification.
Figures



References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. https://doi.org/ 10.1016/S0022-2836(05)80360-2. - DOI - PubMed
-
- Almeida JS. Sequence analysis by iterated maps, a review. Briefings in Bioinformatics. 2013. October;15(3):369–375. 10.1093/bib/bbt072. https://academic.oup.com/bib/article-pdf/15/3/369/450331/bbt072.pdf. - DOI - PMC - PubMed
-
- Lebatteux D, Remita AM, Diallo AB. Toward an Alignment-Free Method for Feature Extraction and Accurate Classification of Viral Sequences. Journal of Computational Biology. 2019;26(6):519–535. https://doi.org/10.1089/cmb.2018.0239. https://doi.org/10.1089/cmb.2018.0239. - DOI - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous