

This is a preprint.
Inferring gene-pathway associations from consolidated transcriptome datasets: an interactive gene network explorer for Tetrahymena thermophila
- PMID: 39713406
- PMCID: PMC11661410
- DOI: 10.1101/2024.12.12.627356
Inferring gene-pathway associations from consolidated transcriptome datasets: an interactive gene network explorer for Tetrahymena thermophila
Update in
-
Inferring gene-pathway associations from consolidated transcriptome datasets: an interactive gene network explorer for Tetrahymena thermophila.NAR Genom Bioinform. 2025 May 27;7(2):lqaf067. doi: 10.1093/nargab/lqaf067. eCollection 2025 Jun. NAR Genom Bioinform. 2025. PMID: 40432793 Free PMC article.
Abstract
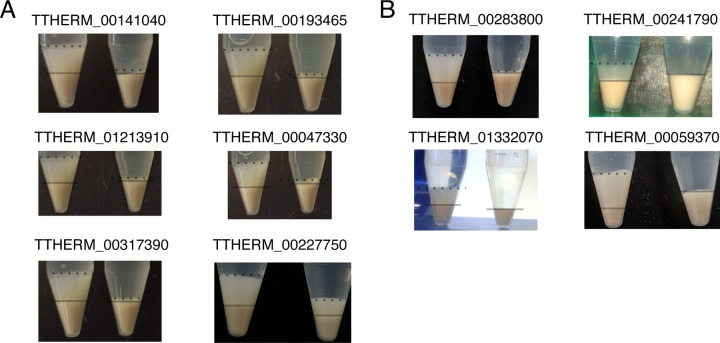
Although an established model organism, Tetrahymena thermophila remains comparatively inaccessible to high throughput screens, and alternative bioinformatic approaches still rely on unconnected datasets and outdated algorithms. Here, we report a new approach to consolidating RNA-seq and microarray data based on a systematic exploration of parameters and computational controls, enabling us to infer functional gene associations from their co-expression patterns. To illustrate the power of this approach, we took advantage of new data regarding a previously studied pathway, the biogenesis of a secretory organelle called the mucocyst. Our untargeted clustering approach recovered over 80% of the genes that were previously verified to play a role in mucocyst biogenesis. Furthermore, we tested four new genes that we predicted to be mucocyst-associated based on their co-expression and found that knocking out each of them results in mucocyst secretion defects. We also found that our approach succeeds in clustering genes associated with several other cellular pathways that we evaluated based on prior literature. We present the Tetrahymena Gene Network Explorer (TGNE) as an interactive tool for genetic hypothesis generation and functional annotation in this organism and as a framework for building similar tools for other systems.
Figures




Similar articles
-
Inferring gene-pathway associations from consolidated transcriptome datasets: an interactive gene network explorer for Tetrahymena thermophila.NAR Genom Bioinform. 2025 May 27;7(2):lqaf067. doi: 10.1093/nargab/lqaf067. eCollection 2025 Jun. NAR Genom Bioinform. 2025. PMID: 40432793 Free PMC article.
-
Whole Genome Sequencing Identifies a Novel Factor Required for Secretory Granule Maturation in Tetrahymena thermophila.G3 (Bethesda). 2016 Aug 9;6(8):2505-16. doi: 10.1534/g3.116.028878. G3 (Bethesda). 2016. PMID: 27317773 Free PMC article.
-
An aspartyl cathepsin, CTH3, is essential for proprotein processing during secretory granule maturation in Tetrahymena thermophila.Mol Biol Cell. 2014 Aug 15;25(16):2444-60. doi: 10.1091/mbc.E14-03-0833. Epub 2014 Jun 18. Mol Biol Cell. 2014. PMID: 24943840 Free PMC article.
-
Tetrahymena as a Unicellular Model Eukaryote: Genetic and Genomic Tools.Genetics. 2016 Jun;203(2):649-65. doi: 10.1534/genetics.114.169748. Genetics. 2016. PMID: 27270699 Free PMC article. Review.
-
Statistical and Machine Learning Approaches to Predict Gene Regulatory Networks From Transcriptome Datasets.Front Plant Sci. 2018 Nov 29;9:1770. doi: 10.3389/fpls.2018.01770. eCollection 2018. Front Plant Sci. 2018. PMID: 30555503 Free PMC article. Review.
References
-
- Jiang C., Gu S., Pan T., Wang X., Qin W., Wang G., Gao X., Zhang J., Chen K., Warren A., et al. (2024) Dynamics and timing of diversification events of ciliated eukaryotes from a large phylogenomic perspective. Molecular Phylogenetics and Evolution, 197, 108110. - PubMed
-
- Kuppannan A., Jiang Y.-Y., Maier W., Liu C., Lang C.F., Cheng C.-Y., Field M.C., Zhao M., Zoltner M. and Turkewitz A.P. (2022) A novel membrane complex is required for docking and regulated exocytosis of lysosome-related organelles in Tetrahymena thermophila. PLOS Genetics, 18, e1010194. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources