

This is a preprint.
MaAsLin 3: Refining and extending generalized multivariable linear models for meta-omic association discovery
- PMID: 39713460
- PMCID: PMC11661281
- DOI: 10.1101/2024.12.13.628459
MaAsLin 3: Refining and extending generalized multivariable linear models for meta-omic association discovery
Abstract
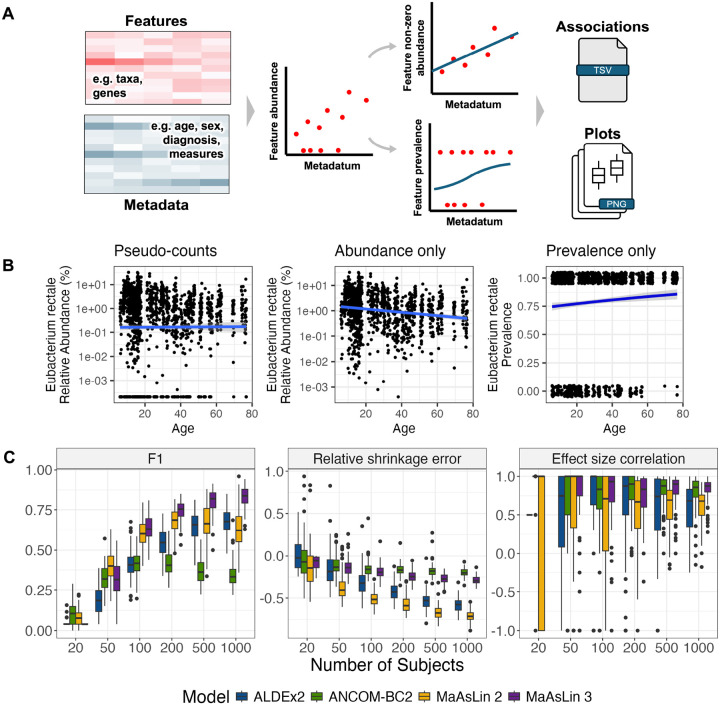
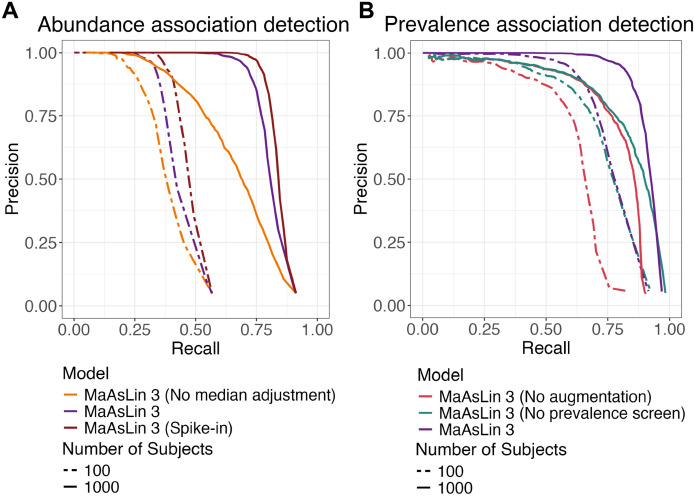
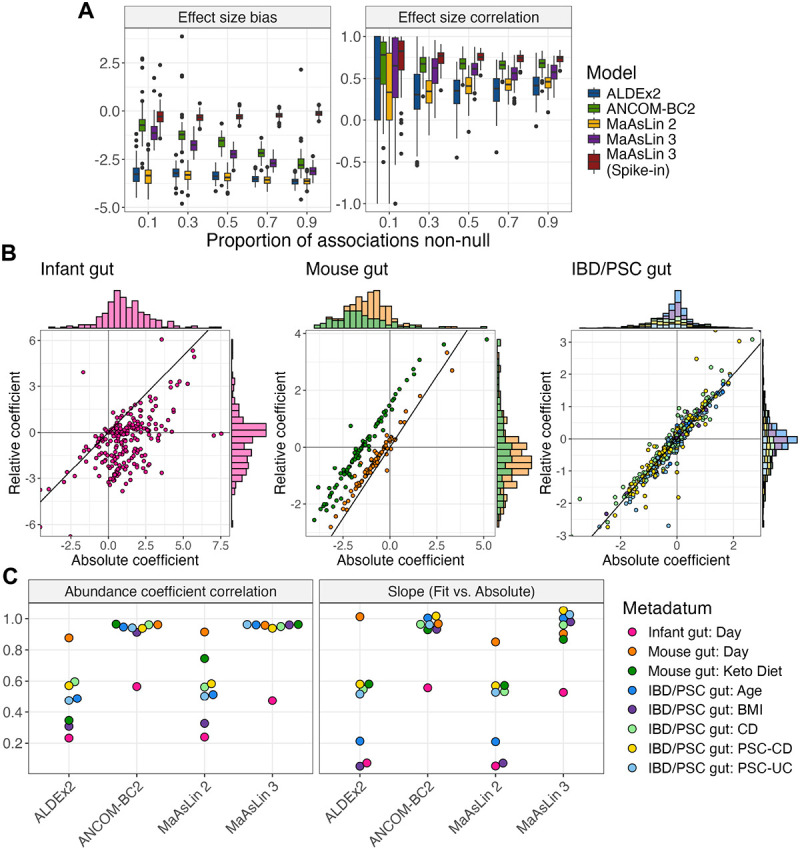
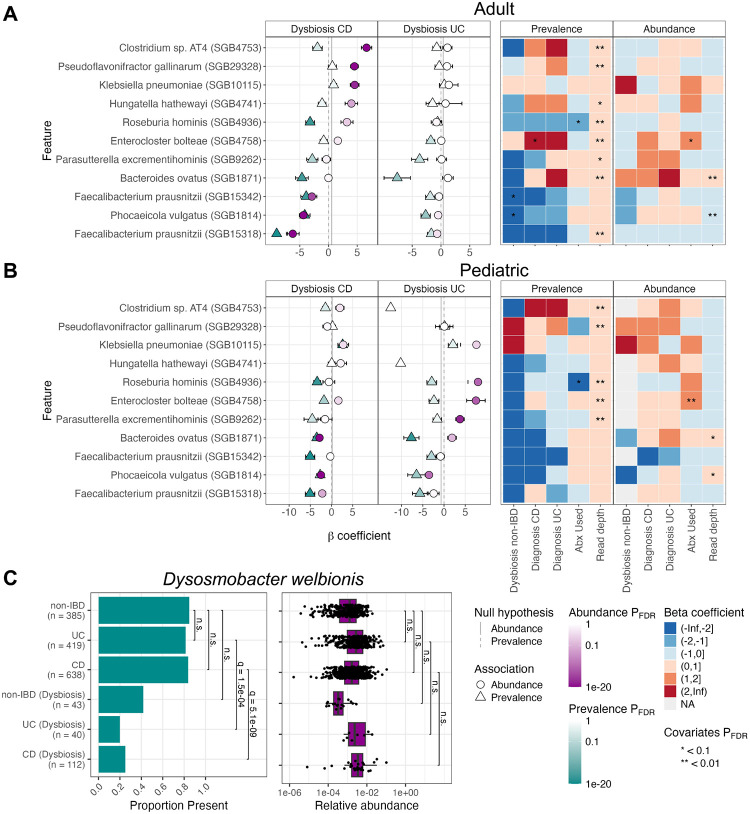
A key question in microbial community analysis is determining which microbial features are associated with community properties such as environmental or health phenotypes. This statistical task is impeded by characteristics of typical microbial community profiling technologies, including sparsity (which can be either technical or biological) and the compositionality imposed by most nucleotide sequencing approaches. Many models have been proposed that focus on how the relative abundance of a feature (e.g. taxon or pathway) relates to one or more covariates. Few of these, however, simultaneously control false discovery rates, achieve reasonable power, incorporate complex modeling terms such as random effects, and also permit assessment of prevalence (presence/absence) associations and absolute abundance associations (when appropriate measurements are available, e.g. qPCR or spike-ins). Here, we introduce MaAsLin 3 (Microbiome Multivariable Associations with Linear Models), a modeling framework that simultaneously identifies both abundance and prevalence relationships in microbiome studies with modern, potentially complex designs. MaAsLin 3 also newly accounts for compositionality with experimental (spike-ins and total microbial load estimation) or computational techniques, and it expands the space of biological hypotheses that can be tested with inference for new covariate types. On a variety of synthetic and real datasets, MaAsLin 3 outperformed current state-of-the-art differential abundance methods in testing and inferring associations from compositional data. When applied to the Inflammatory Bowel Disease Multi-omics Database, MaAsLin 3 corroborated many previously reported microbial associations with the inflammatory bowel diseases, but notably 77% of associations were with feature prevalence rather than abundance. In summary, MaAsLin 3 enables researchers to identify microbiome associations with higher accuracy and more specific association types, especially in complex datasets with multiple covariates and repeated measures.
Conflict of interest statement
Competing interests C.H. declares the following associations: Seres Therapeutics (scientific advisory board, microbiome therapies), Microbiome Insights (scientific advisory board, microbiome data generation), Zoe (scientific advisory board), Empress (scientific advisory board, microbiome therapies).
Figures
References
-
- Gloor G. B., Wu J. R., Pawlowsky-Glahn V. & Egozcue J. J. It’s all relative: analyzing microbiome data as compositions. Annals of Epidemiology 26, 322–329 (2016). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources