

Stabilization of mitochondria-associated endoplasmic reticulum membranes regulates Aβ generation in a three-dimensional neural model of Alzheimer's disease
- PMID: 39713841
- PMCID: PMC11848173
- DOI: 10.1002/alz.14417
Stabilization of mitochondria-associated endoplasmic reticulum membranes regulates Aβ generation in a three-dimensional neural model of Alzheimer's disease
Abstract
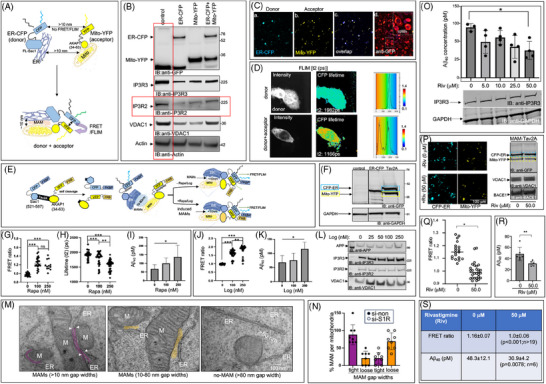
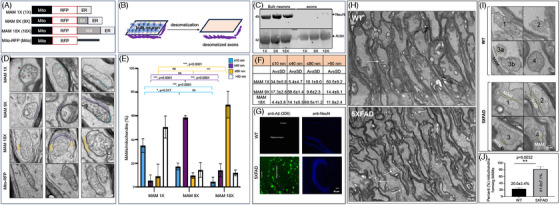
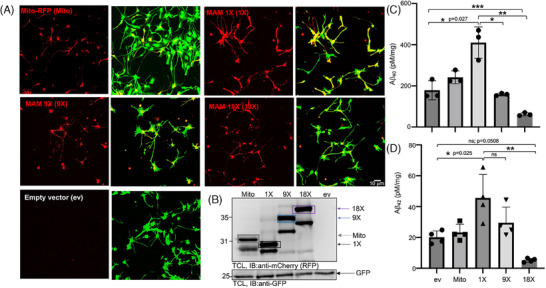
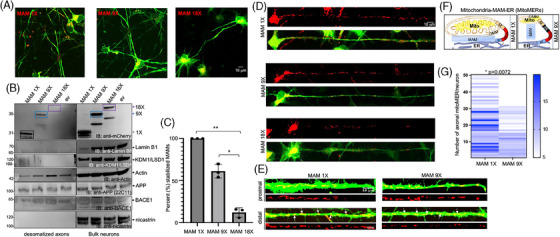
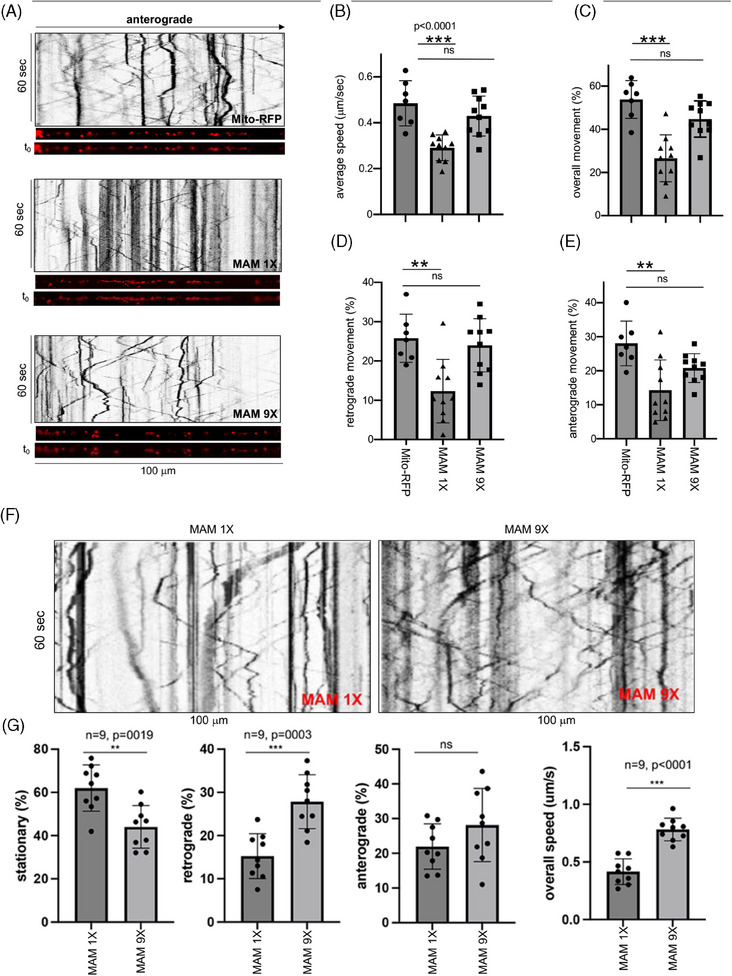
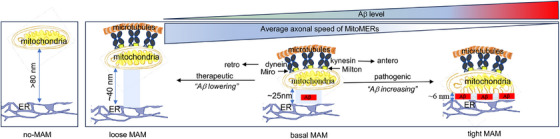
Introduction: We previously demonstrated that regulating mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) affects axonal Aβ generation in a well-characterized three-dimensional (3D) neural Alzheimer's disease (AD) model. MAMs vary in thickness and length, impacting their functions. Here, we examined the effect of MAM thickness on Aβ in our 3D neural model of AD.
Methods: We employed fluorescence resonance energy transfer (FRET) or fluorescence-based MAM stabilizers, electron microscopy, Aβ enzyme-linked immunosorbent assay (ELISA), and live-cell imaging with kymography to assess how stabilizing MAMs of different gap widths influence Aβ production and MAM axonal mobility.
Results: Stabilizing tight MAMs (∼6 nm gap width) significantly increased Aβ levels, whereas basal (∼25 nm) and loose MAMs (∼40 nm) maintained or reduced Aβ levels, respectively. Tight MAMs reduced mitochondrial axonal velocity compared to basal MAMs, while loose MAMs showed severely reduced axonal distribution.
Discussion: Our findings suggest that stabilizing MAMs of specific gap widths, particularly in axons, without complete destabilization could be an effective therapeutic strategy for AD.
Highlights: The stabilization of MAMs exacerbates or ameliorates Aβ generation from AD neurons in a MAM gap width-dependent manner. A specific stabilization threshold within the MAM gap width spectrum shifts the amyloidogenic process to non-amyloidogenic. Tight MAMs slow down mitochondrial axonal transport compared to lose MAMs offering a quantitative method for measuring MAM stabilization.
Keywords: Alzheimer's disease; amyloid β; endoplasmic reticulum; mitochondria; mitochondria‐associated ER membranes; β‐amyloid precursor protein; β‐site APP cleaving enzyme.
© 2024 The Author(s). Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
All authors report no conflict of interest. Author disclosures are available in the Supporting Information.
Figures
Similar articles
-
Quantitative Analysis of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Stabilization in a Neural Model of Alzheimer's Disease (AD).J Vis Exp. 2025 Jan 10;(215). doi: 10.3791/66129. J Vis Exp. 2025. PMID: 39878941
-
Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease.EMBO J. 2017 Nov 15;36(22):3356-3371. doi: 10.15252/embj.201796797. Epub 2017 Oct 10. EMBO J. 2017. PMID: 29018038 Free PMC article.
-
Axonal generation of amyloid-β from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes.Cell Rep. 2021 May 18;35(7):109134. doi: 10.1016/j.celrep.2021.109134. Cell Rep. 2021. PMID: 34010653 Free PMC article.
-
The correlation between mitochondria-associated endoplasmic reticulum membranes (MAMs) and Ca2+ transport in the pathogenesis of diseases.Acta Pharmacol Sin. 2025 Feb;46(2):271-291. doi: 10.1038/s41401-024-01359-9. Epub 2024 Aug 8. Acta Pharmacol Sin. 2025. PMID: 39117969 Review.
-
Soluble and insoluble protein aggregates, endoplasmic reticulum stress, and vascular dysfunction in Alzheimer's disease and cardiovascular diseases.Geroscience. 2023 Jun;45(3):1411-1438. doi: 10.1007/s11357-023-00748-y. Epub 2023 Feb 24. Geroscience. 2023. PMID: 36823398 Free PMC article. Review.
Cited by
-
Endoplasmic reticulum-Mitochondria Coupling in Alzheimer's Disease.Contact (Thousand Oaks). 2025 Jul 17;8:25152564251330069. doi: 10.1177/25152564251330069. eCollection 2025 Jan-Dec. Contact (Thousand Oaks). 2025. PMID: 40689264 Free PMC article. Review.
-
Mitochondria-Associated Endoplasmic Reticulum Membranes in Human Health and Diseases.MedComm (2020). 2025 Jun 27;6(7):e70259. doi: 10.1002/mco2.70259. eCollection 2025 Jul. MedComm (2020). 2025. PMID: 40584408 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
